

Deep cell phenotyping and spatial analysis of multiplexed imaging with TRACERx-PHLEX
- PMID: 38879602
- PMCID: PMC11180132
- DOI: 10.1038/s41467-024-48870-5
Deep cell phenotyping and spatial analysis of multiplexed imaging with TRACERx-PHLEX
Abstract
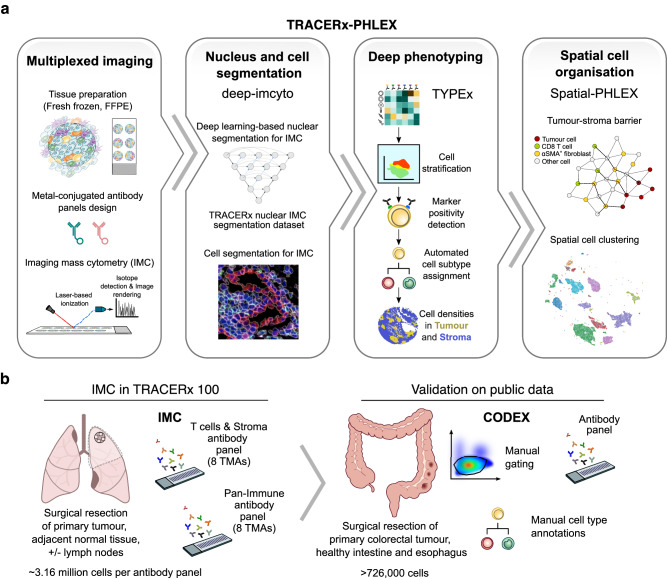
The growing scale and dimensionality of multiplexed imaging require reproducible and comprehensive yet user-friendly computational pipelines. TRACERx-PHLEX performs deep learning-based cell segmentation (deep-imcyto), automated cell-type annotation (TYPEx) and interpretable spatial analysis (Spatial-PHLEX) as three independent but interoperable modules. PHLEX generates single-cell identities, cell densities within tissue compartments, marker positivity calls and spatial metrics such as cellular barrier scores, along with summary graphs and spatial visualisations. PHLEX was developed using imaging mass cytometry (IMC) in the TRACERx study, validated using published Co-detection by indexing (CODEX), IMC and orthogonal data and benchmarked against state-of-the-art approaches. We evaluated its use on different tissue types, tissue fixation conditions, image sizes and antibody panels. As PHLEX is an automated and containerised Nextflow pipeline, manual assessment, programming skills or pathology expertise are not essential. PHLEX offers an end-to-end solution in a growing field of highly multiplexed data and provides clinically relevant insights.
© 2024. The Author(s).
Conflict of interest statement
C.S. acknowledges grant support from Bristol Myers Squibb related to this work and grants from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche-Ventana, Invitae, Ono Pharmaceutical, and Personalis outside of the submitted work. He is Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board. During the conduct of the study outside the submitted work, C.S. has received consultant fees from Achilles Therapeutics (also a SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, China Innovation Centre of Roche (CICoR) formerly Roche Innovation Centre—Shanghai, Metabomed (until July 2022), Relay Therapeutics SAB member, Saga Diagnostics SAB member and the Sarah Cannon Research Institute. Outside of the submitted work, C.S. has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer, Medixci, and Roche-Ventana. C.S. has previously held stock options in Apogen Biotechnologies and GRAIL, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, Relay Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. declares a patent application (PCT/US2017/028013) for methods for lung cancer; targeting neoantigens (PCT/EP2016/059401); identifying patient response to immune checkpoint blockade (PCT/EP2016/071471); methods for lung cancer detection (US20190106751A1); identifying patients who respond to cancer treatment (PCT/GB2018/051912); determining HLA LOH (PCT/GB2018/052004); predicting survival rates of patients with cancer (PCT/GB2020/050221); methods for systems and tumour monitoring (PCT/EP2022/077987). C.S. is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumour recurrence. This patent has been licensed to a commercial entity and under their terms of employment C.S. is due a revenue share of any revenue generated from such license(s). M.A. is a co-inventor on a European patent application (PCT/EP2020/059272) about methods for predicting and preventing cancer in patients with premalignant lesions. J.D. reports grants, personal fees, and nonfinancial support from AstraZeneca, personal fees from Bayer, Jubilant, Theras, BridgeBio, Vividion, Novartis, and grants and nonfinancial support from Bristol Myers Squibb and Revolution Medicines, outside the submitted work. E.S. has received funded research agreements from Merck Sharp Dohme, AstraZeneca and personal fees from Phenomic outside the submitted work. C.T.H. has received speaker fees from AstraZeneca, holds a paid advisory role for GenesisCare UK, research funding and support from Roche, AstraZeneca and Personalis. S.A.Q. reports other support from Achilles Therapeutics, grants from Roche, and Sairoopa outside the submitted work. J.L.R. reports speaker fees from Boehringer Ingelheim and GlaxoSmithKline, consults for Achilles Therapeutics Ltd and has filed patents for cancer early detection (PCT/EP2023/076521 and PCT/EP2023/076511). D.A.M. reports speaker fees from AstraZeneca and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, MIM Software, Bristol Myers Squibb and Eli Lilly and educational support from Takeda and Amgen. The remaining authors declare no competing interests.
Figures






References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
