

Novel formylpeptide receptor 1/2 agonist limits hypertension-induced cardiovascular damage
- PMID: 38879891
- PMCID: PMC11416058
- DOI: 10.1093/cvr/cvae103
Novel formylpeptide receptor 1/2 agonist limits hypertension-induced cardiovascular damage
Abstract
Aims: Formylpeptide receptors (FPRs) play a critical role in the regulation of inflammation, an important driver of hypertension-induced end-organ damage. We have previously reported that the biased FPR small-molecule agonist, compound17b (Cmpd17b), is cardioprotective against acute, severe inflammatory insults. Here, we reveal the first compelling evidence of the therapeutic potential of this novel FPR agonist against a longer-term, sustained inflammatory insult, i.e. hypertension-induced end-organ damage. The parallels between the murine and human hypertensive proteome were also investigated.
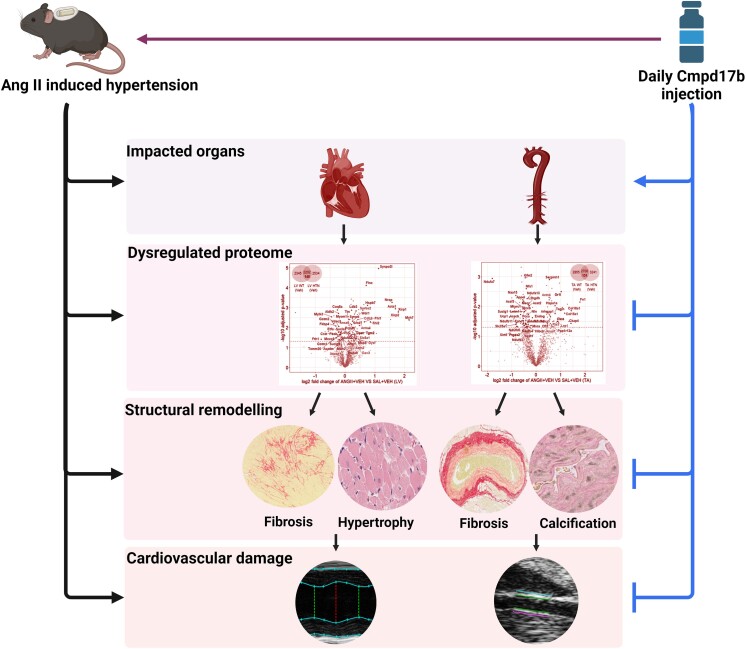
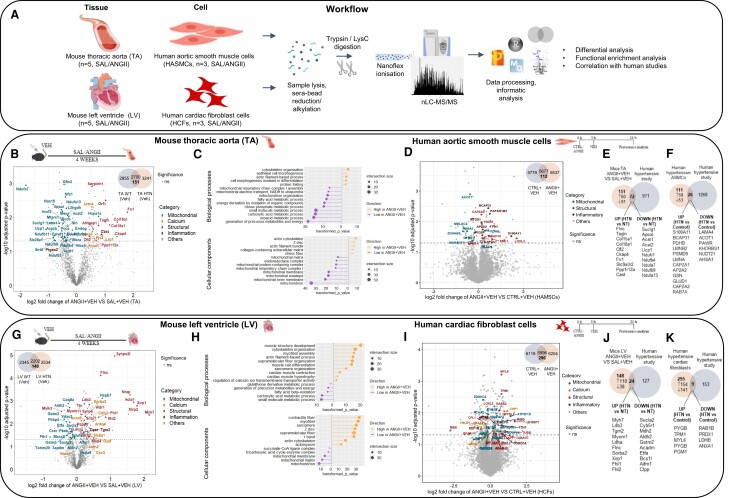
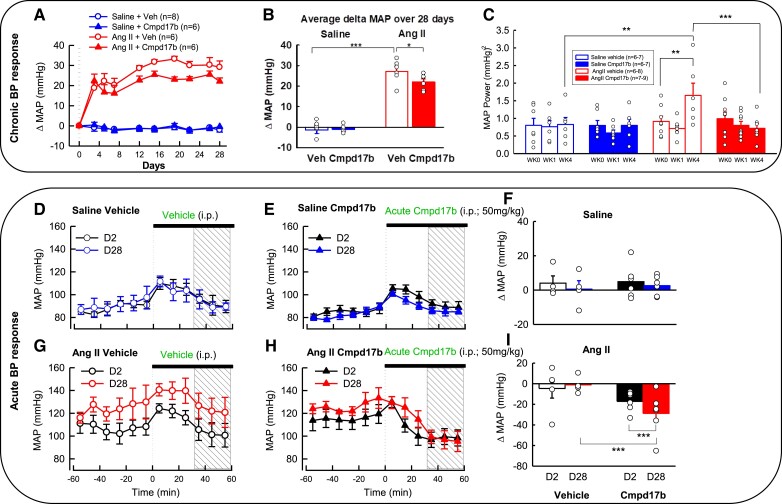
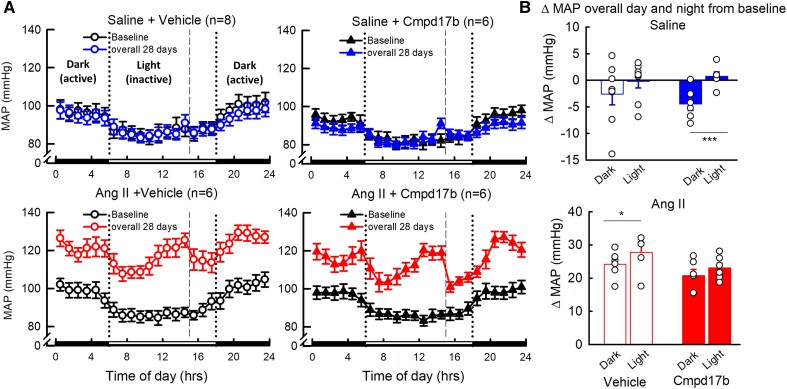
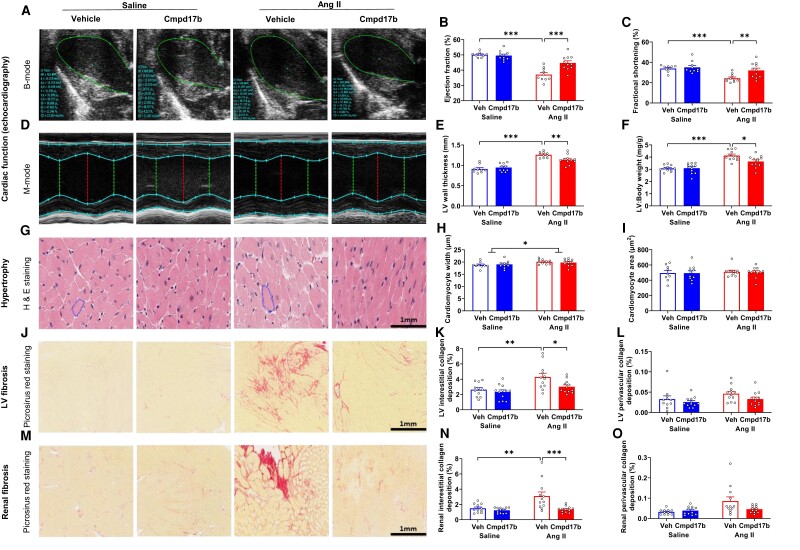
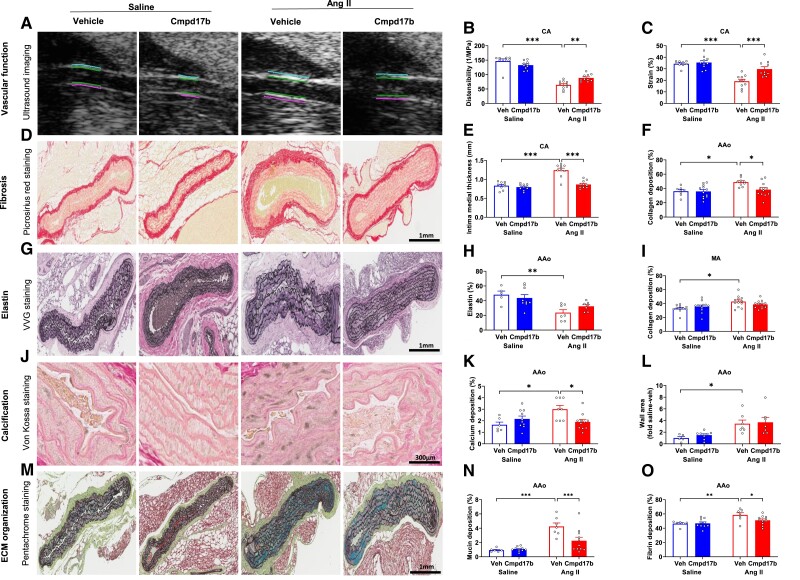
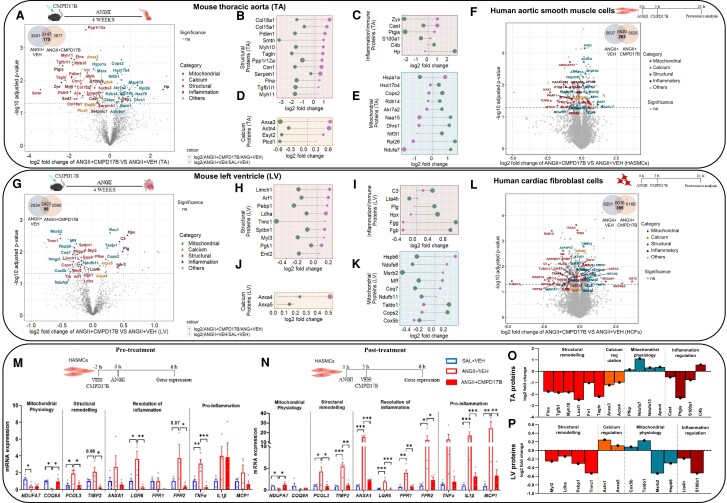
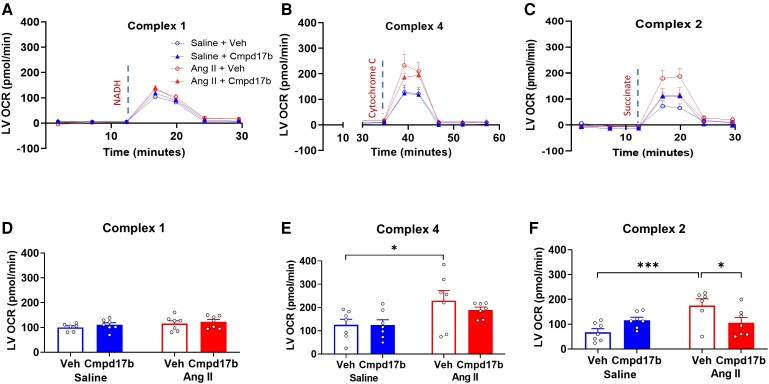
Methods and results: The hypertensive response to angiotensin II (Ang II, 0.7 mg/kg/day, s.c.) was attenuated by Cmpd17b (50 mg/kg/day, i.p.). Impairments in cardiac and vascular function assessed via echocardiography were improved by Cmpd17b in hypertensive mice. This functional improvement was accompanied by reduced cardiac and aortic fibrosis and vascular calcification. Cmpd17b also attenuated Ang II-induced increased cardiac mitochondrial complex 2 respiration. Proteomic profiling of cardiac and aortic tissues and cells, using label-free nano-liquid chromatography with high-sensitivity mass spectrometry, detected and quantified ∼6000 proteins. We report hypertension-impacted protein clusters associated with dysregulation of inflammatory, mitochondrial, and calcium responses, as well as modified networks associated with cardiovascular remodelling, contractility, and structural/cytoskeletal organization. Cmpd17b attenuated hypertension-induced dysregulation of multiple proteins in mice, and of these, ∼110 proteins were identified as similarly dysregulated in humans suffering from adverse aortic remodelling and cardiac hypertrophy.
Conclusion: We have demonstrated, for the first time, that the FPR agonist Cmpd17b powerfully limits hypertension-induced end-organ damage, consistent with proteome networks, supporting development of pro-resolution FPR-based therapeutics for treatment of systemic hypertension complications.
Trial registration: ClinicalTrials.gov NCT03335553.
Keywords: Angiotensin II; Compound17b; End-organ damage; Formylpeptide receptors; Hypertension; Proteomics.
© The Author(s) 2024. Published by Oxford University Press on behalf of the European Society of Cardiology.
Conflict of interest statement
Conflict of interest: none declared.
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
