

Data-Independent Acquisition: A Milestone and Prospect in Clinical Mass Spectrometry-Based Proteomics
- PMID: 38880244
- PMCID: PMC11380018
- DOI: 10.1016/j.mcpro.2024.100800
Data-Independent Acquisition: A Milestone and Prospect in Clinical Mass Spectrometry-Based Proteomics
Abstract
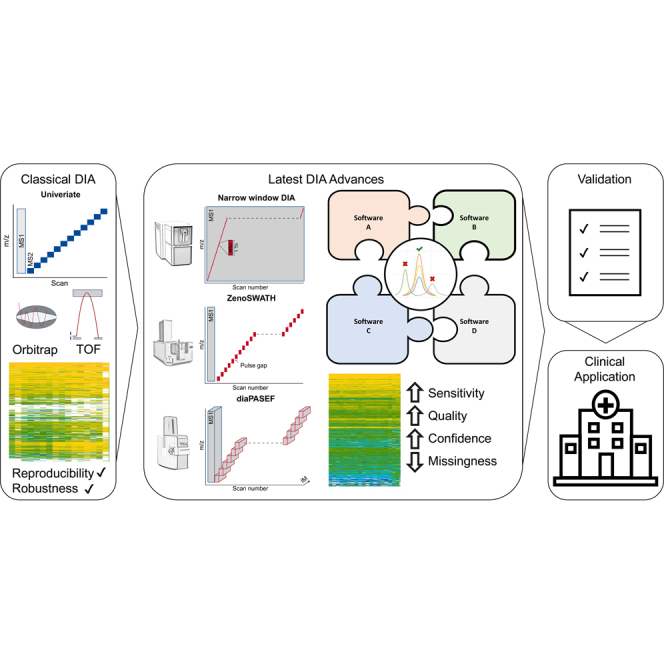
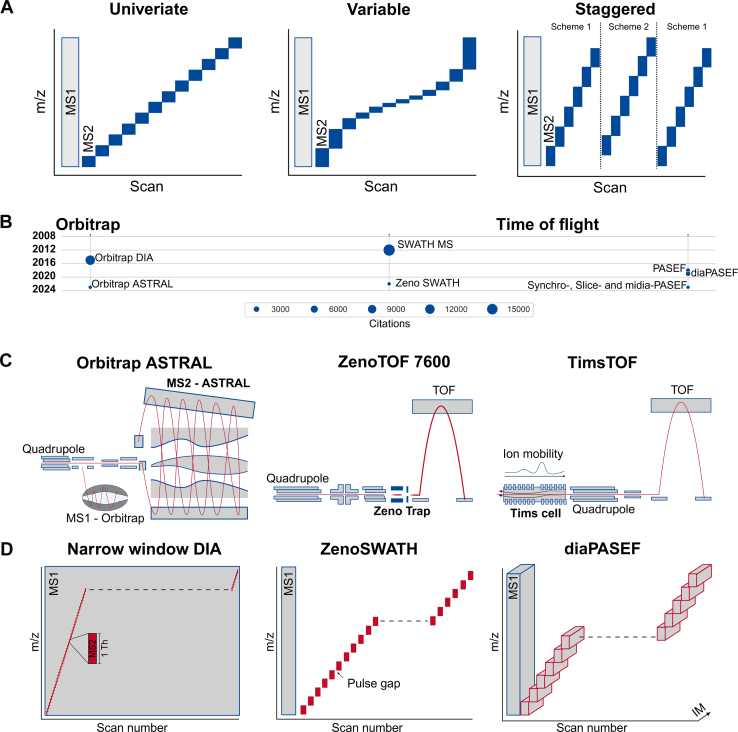
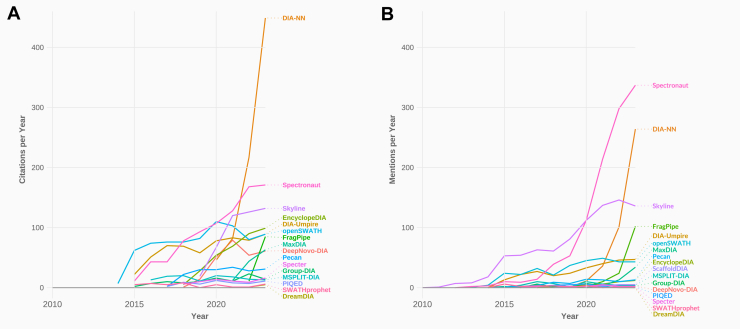
Data-independent acquisition (DIA) has revolutionized the field of mass spectrometry (MS)-based proteomics over the past few years. DIA stands out for its ability to systematically sample all peptides in a given m/z range, allowing an unbiased acquisition of proteomics data. This greatly mitigates the issue of missing values and significantly enhances quantitative accuracy, precision, and reproducibility compared to many traditional methods. This review focuses on the critical role of DIA analysis software tools, primarily focusing on their capabilities and the challenges they address in proteomic research. Advances in MS technology, such as trapped ion mobility spectrometry, or high field asymmetric waveform ion mobility spectrometry require sophisticated analysis software capable of handling the increased data complexity and exploiting the full potential of DIA. We identify and critically evaluate leading software tools in the DIA landscape, discussing their unique features, and the reliability of their quantitative and qualitative outputs. We present the biological and clinical relevance of DIA-MS and discuss crucial publications that paved the way for in-depth proteomic characterization in patient-derived specimens. Furthermore, we provide a perspective on emerging trends in clinical applications and present upcoming challenges including standardization and certification of MS-based acquisition strategies in molecular diagnostics. While we emphasize the need for continuous development of software tools to keep pace with evolving technologies, we advise researchers against uncritically accepting the results from DIA software tools. Each tool may have its own biases, and some may not be as sensitive or reliable as others. Our overarching recommendation for both researchers and clinicians is to employ multiple DIA analysis tools, utilizing orthogonal analysis approaches to enhance the robustness and reliability of their findings.
Keywords: clinical proteomics; data-independent acquisition; mass spectrometry.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare no competing interests.
Figures
References
-
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Tang W.H., Halpern B.R., Shilov I.V., Seymour S.L., Keating S.P., Loboda A., et al. Discovering known and unanticipated protein modifications using MS/MS database searching. Anal. Chem. 2005;77:3931–3946. - PubMed
-
- Purvine S., Eppel J.-T., Yi E.C., Goodlett D.R. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3:847–850. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
