

This is a preprint.
ACK1 and BRK non-receptor tyrosine kinase deficiencies are associated with familial systemic lupus and involved in efferocytosis
- PMID: 38883731
- PMCID: PMC11177913
- DOI: 10.1101/2024.02.15.24302255
ACK1 and BRK non-receptor tyrosine kinase deficiencies are associated with familial systemic lupus and involved in efferocytosis
Update in
-
ACK1 and BRK non-receptor tyrosine kinase deficiencies are associated with familial systemic lupus and involved in efferocytosis.Elife. 2024 Nov 21;13:RP96085. doi: 10.7554/eLife.96085. Elife. 2024. PMID: 39570652 Free PMC article.
Abstract
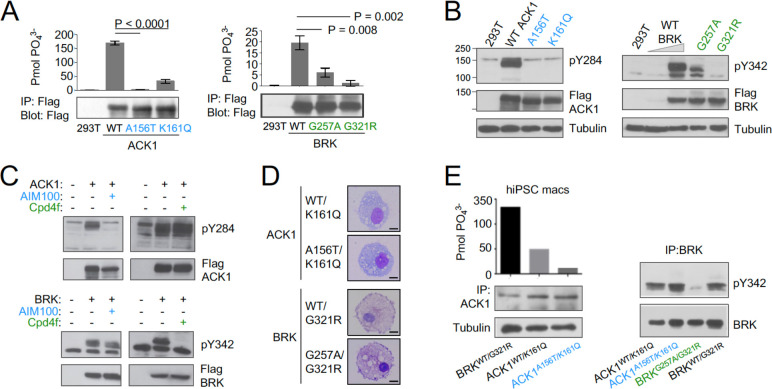
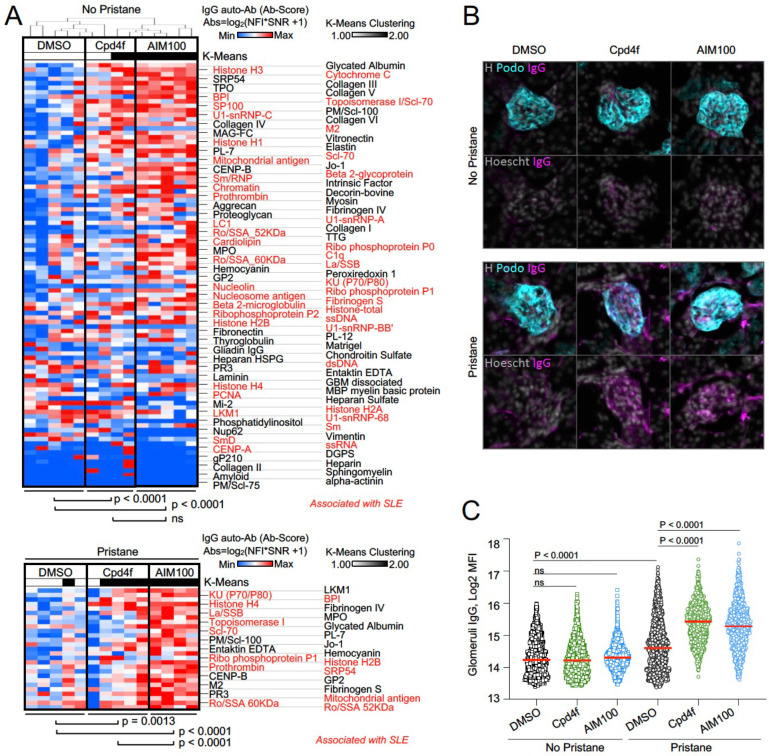
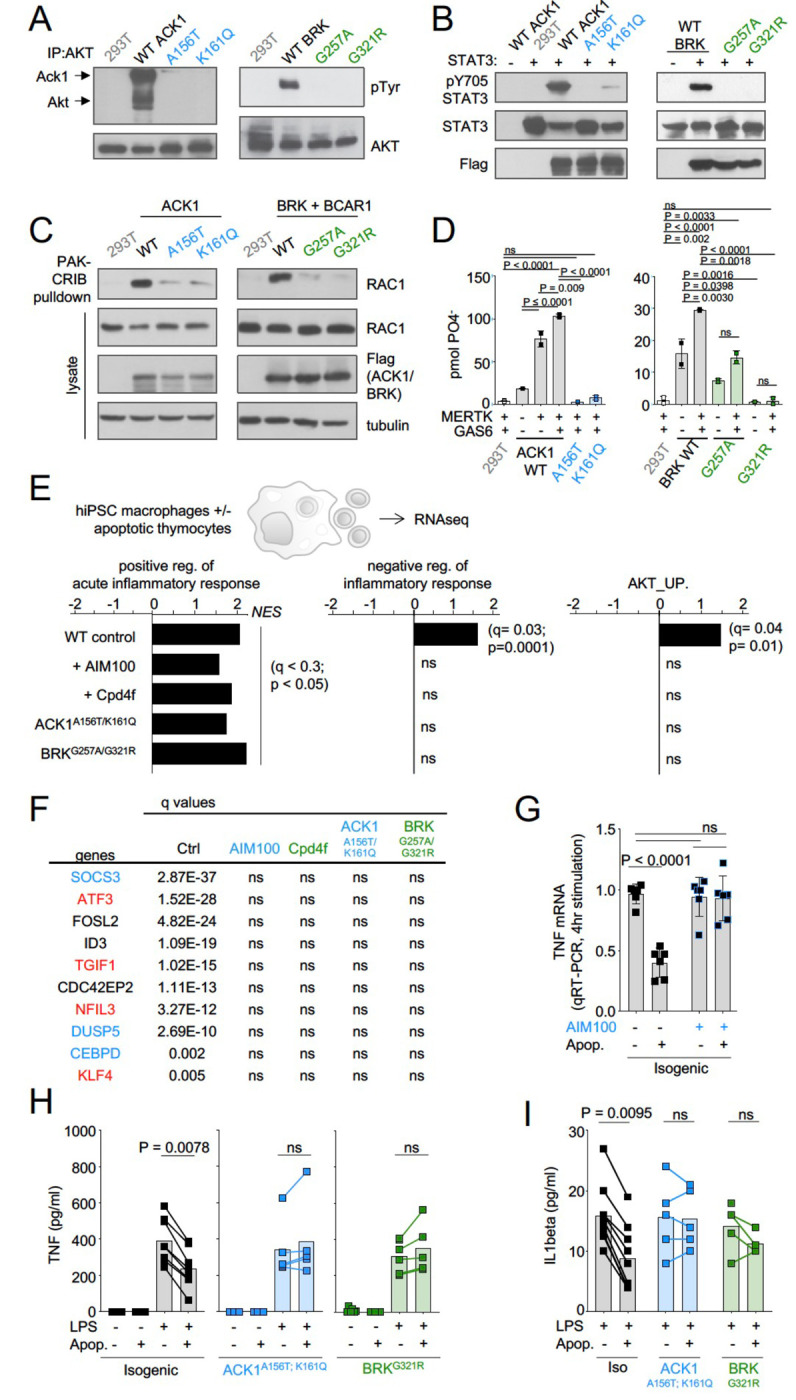
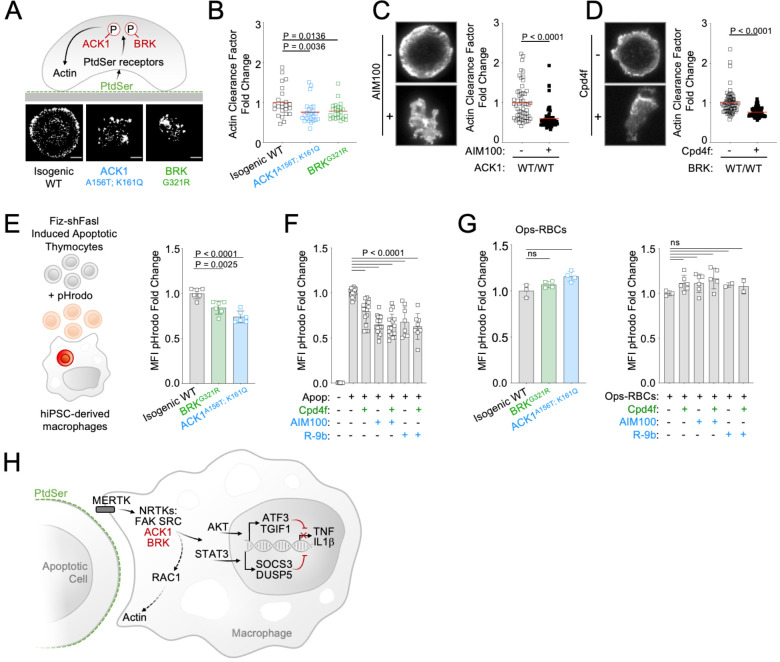
Systemic Lupus Erythematosus (SLE) is an autoimmune disease, the pathophysiology and genetic basis of which are incompletely understood. Using a forward genetic screen in multiplex families with systemic lupus erythematosus (SLE) we identified an association between SLE and compound heterozygous deleterious variants in the non-receptor tyrosine kinases (NRTKs) ACK1 and BRK. Experimental blockade of ACK1 or BRK increased circulating autoantibodies in vivo in mice and exacerbated glomerular IgG deposits in an SLE mouse model. Mechanistically, non-receptor tyrosine kinases (NRTKs) regulate activation, migration, and proliferation of immune cells. We found that the patients' ACK1 and BRK variants impair efferocytosis, the MERTK-mediated anti-inflammatory response to apoptotic cells, in human induced Pluripotent Stem Cell (hiPSC)-derived macrophages, which may contribute to SLE pathogenesis. Overall, our data suggest that ACK1 and BRK deficiencies are associated with human SLE and impair efferocytosis in macrophages.
Figures

References
-
- Dall’Era M. in Current Rheumatology Diagnosis and Treatment. 3rd ed. (eds Imboden J.B., Hellman D.B., & Stone J.H.) (McGraw-Hill, 2013).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous