

The SMC5/6 complex prevents genotoxicity upon APOBEC3A-mediated replication stress
- PMID: 38886582
- PMCID: PMC11294446
- DOI: 10.1038/s44318-024-00137-x
The SMC5/6 complex prevents genotoxicity upon APOBEC3A-mediated replication stress
Abstract
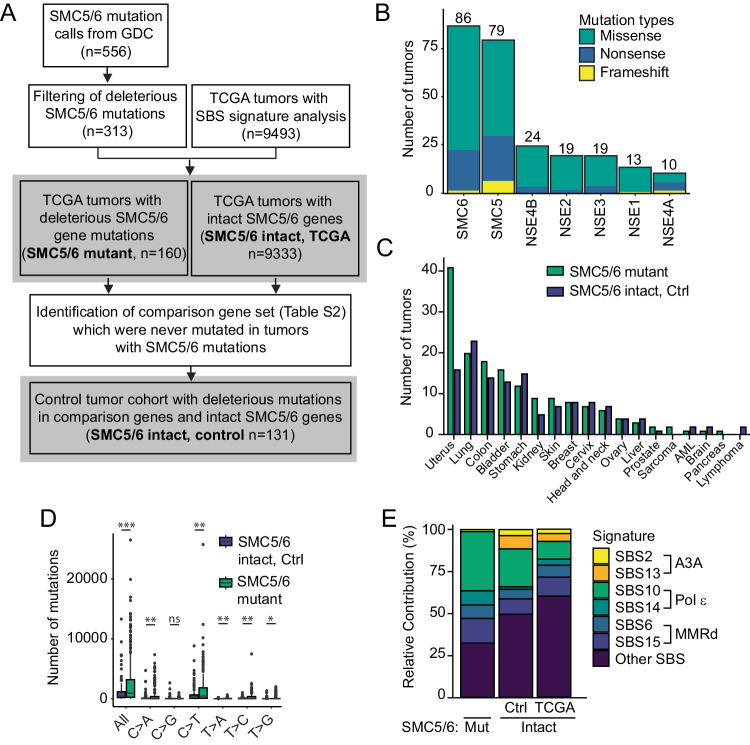
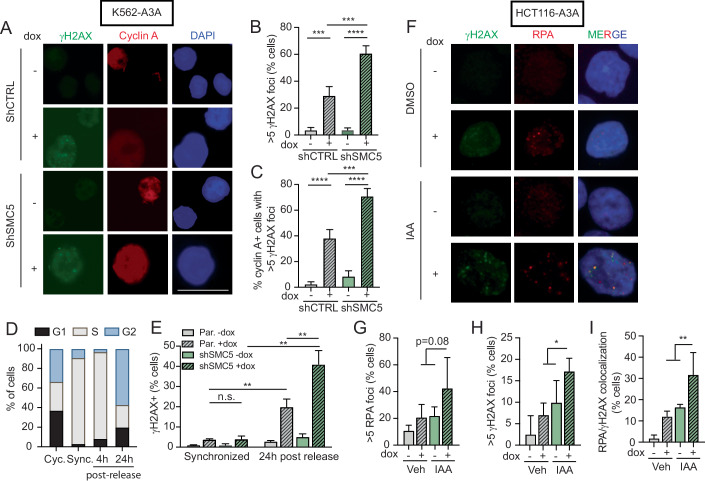
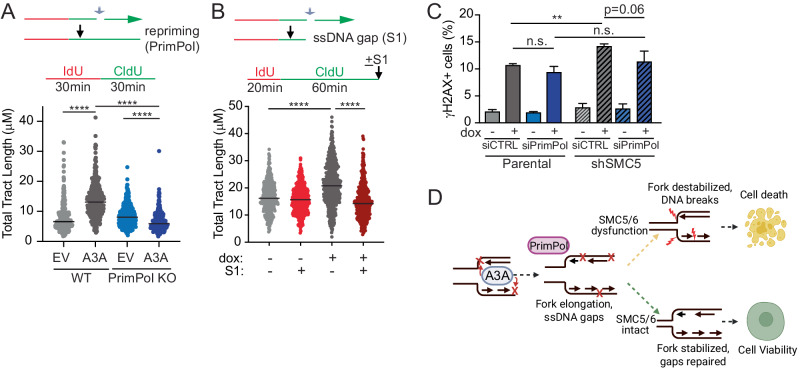
Mutational patterns caused by APOBEC3 cytidine deaminase activity are evident throughout human cancer genomes. In particular, the APOBEC3A family member is a potent genotoxin that causes substantial DNA damage in experimental systems and human tumors. However, the mechanisms that ensure genome stability in cells with active APOBEC3A are unknown. Through an unbiased genome-wide screen, we define the Structural Maintenance of Chromosomes 5/6 (SMC5/6) complex as essential for cell viability when APOBEC3A is active. We observe an absence of APOBEC3A mutagenesis in human tumors with SMC5/6 dysfunction, consistent with synthetic lethality. Cancer cells depleted of SMC5/6 incur substantial genome damage from APOBEC3A activity during DNA replication. Further, APOBEC3A activity results in replication tract lengthening which is dependent on PrimPol, consistent with re-initiation of DNA synthesis downstream of APOBEC3A-induced lesions. Loss of SMC5/6 abrogates elongated replication tracts and increases DNA breaks upon APOBEC3A activity. Our findings indicate that replication fork lengthening reflects a DNA damage response to APOBEC3A activity that promotes genome stability in an SMC5/6-dependent manner. Therefore, SMC5/6 presents a potential therapeutic vulnerability in tumors with active APOBEC3A.
Keywords: Cancer Mutagenesis; Cytidine Deaminase; Genome Integrity; Mutational Signatures; Replication Stress.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Update of
-
The SMC5/6 complex prevents genotoxicity upon APOBEC3A-mediated replication stress.bioRxiv [Preprint]. 2024 Mar 24:2023.11.28.568952. doi: 10.1101/2023.11.28.568952. bioRxiv. 2024. Update in: EMBO J. 2024 Aug;43(15):3240-3255. doi: 10.1038/s44318-024-00137-x. PMID: 38077016 Free PMC article. Updated. Preprint.
References
-
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A (2014) Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16:281–293 - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
