

Mitochondrial calcium in cardiac ischemia/reperfusion injury and cardioprotection
- PMID: 38890208
- PMCID: PMC11319510
- DOI: 10.1007/s00395-024-01060-2
Mitochondrial calcium in cardiac ischemia/reperfusion injury and cardioprotection
Abstract
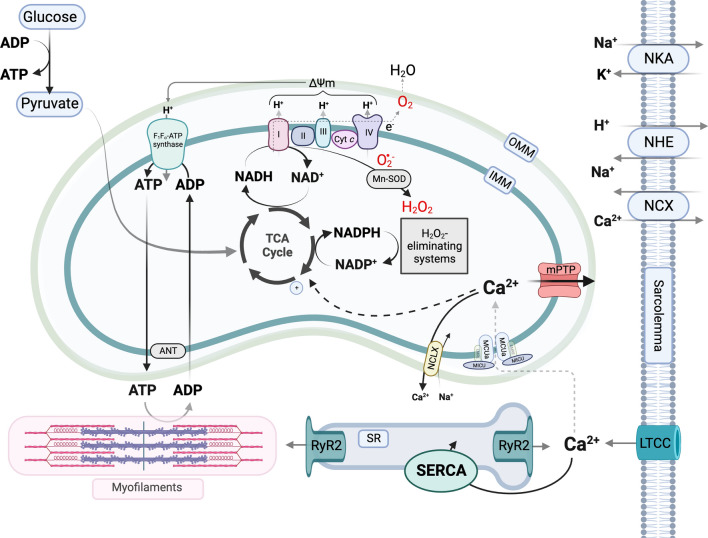
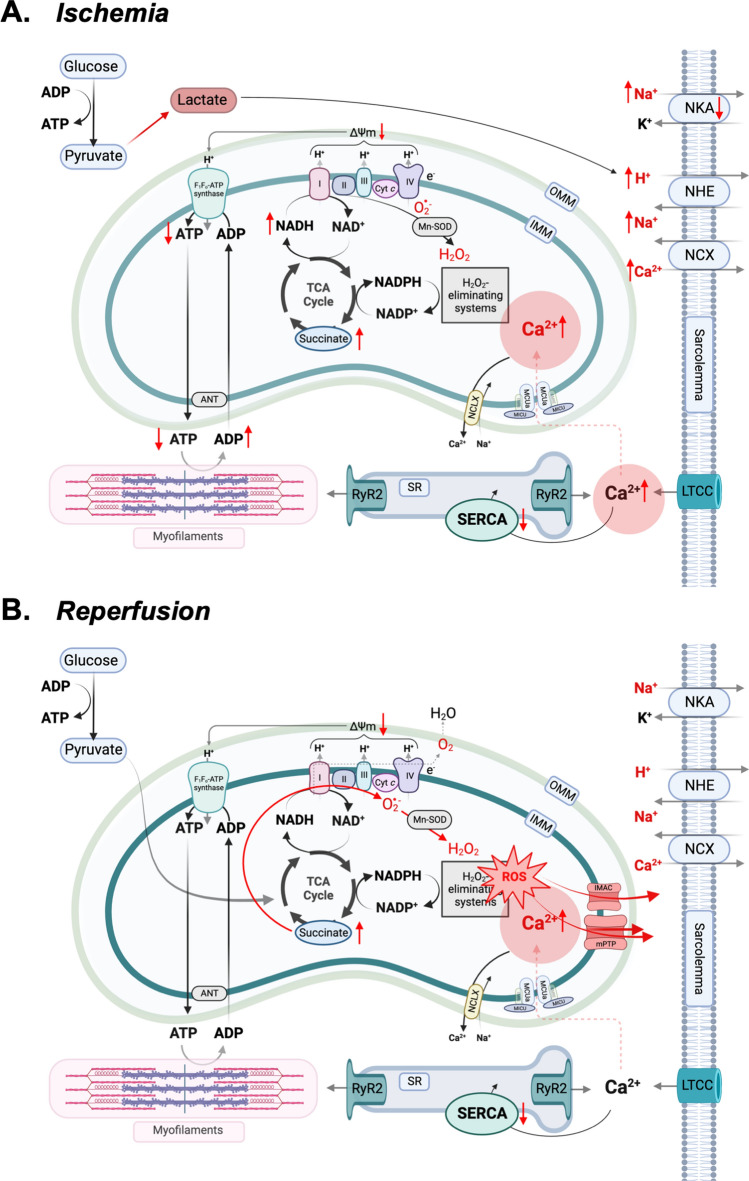
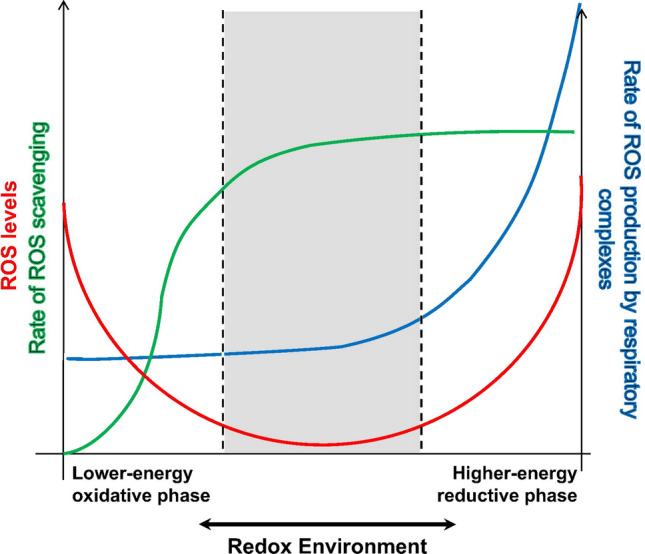
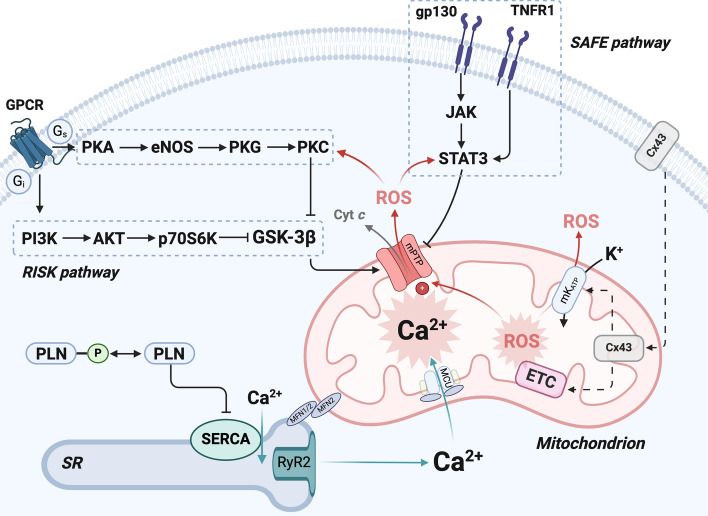
Mitochondrial calcium (Ca2+) signals play a central role in cardiac homeostasis and disease. In the healthy heart, mitochondrial Ca2+ levels modulate the rate of oxidative metabolism to match the rate of adenosine triphosphate consumption in the cytosol. During ischemia/reperfusion (I/R) injury, pathologically high levels of Ca2+ in the mitochondrial matrix trigger the opening of the mitochondrial permeability transition pore, which releases solutes and small proteins from the matrix, causing mitochondrial swelling and ultimately leading to cell death. Pharmacological and genetic approaches to tune mitochondrial Ca2+ handling by regulating the activity of the main Ca2+ influx and efflux pathways, i.e., the mitochondrial Ca2+ uniporter and sodium/Ca2+ exchanger, represent promising therapeutic strategies to protect the heart from I/R injury.
Keywords: Calcium handling; Cardiac myocytes; Ischemia/reperfusion injury; Mitochondria; Myocardial infarction; Reactive oxygen species.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no conflict of interest related to this work.
Figures
Similar articles
-
MCUb Induction Protects the Heart From Postischemic Remodeling.Circ Res. 2020 Jul 17;127(3):379-390. doi: 10.1161/CIRCRESAHA.119.316369. Epub 2020 Apr 17. Circ Res. 2020. PMID: 32299299 Free PMC article.
-
Inhibition of the mPTP and Lipid Peroxidation Is Additively Protective Against I/R Injury.Circ Res. 2024 May 10;134(10):1292-1305. doi: 10.1161/CIRCRESAHA.123.323882. Epub 2024 Apr 15. Circ Res. 2024. PMID: 38618716 Free PMC article.
-
Cardioprotective effect of hyperthyroidism on the stunned rat heart during ischaemia-reperfusion: energetics and role of mitochondria.Exp Physiol. 2015 Jun;100(6):680-97. doi: 10.1113/EP085063. Exp Physiol. 2015. PMID: 25854703
-
How does mitochondrial Ca2+ change during ischemia and reperfusion? Implications for activation of the permeability transition pore.J Gen Physiol. 2025 Jan 6;157(1):e202313520. doi: 10.1085/jgp.202313520. Epub 2024 Dec 19. J Gen Physiol. 2025. PMID: 39699565 Review.
-
Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection.Cell Physiol Biochem. 2007;20(1-4):1-22. doi: 10.1159/000103747. Cell Physiol Biochem. 2007. PMID: 17595511 Review.
Cited by
-
Important Role of Mitochondrial Dysfunction in Immune Triggering and Inflammatory Response in Rheumatoid Arthritis.J Inflamm Res. 2024 Dec 27;17:11631-11657. doi: 10.2147/JIR.S499473. eCollection 2024. J Inflamm Res. 2024. PMID: 39741752 Free PMC article. Review.
-
Role of mitochondrial Ca2+ in stroke: From molecular mechanism to treatment strategy (Review).Mol Med Rep. 2025 Oct;32(4):271. doi: 10.3892/mmr.2025.13636. Epub 2025 Aug 1. Mol Med Rep. 2025. PMID: 40747662 Free PMC article. Review.
-
CHAC1 Mediates Endoplasmic Reticulum Stress-Dependent Ferroptosis in Calcium Oxalate Kidney Stone Formation.Adv Sci (Weinh). 2025 Mar;12(10):e2403992. doi: 10.1002/advs.202403992. Epub 2025 Jan 21. Adv Sci (Weinh). 2025. PMID: 39836526 Free PMC article.
-
FL3 mitigates cardiac ischemia-reperfusion injury by promoting mitochondrial fusion to restore calcium homeostasis.Cell Death Discov. 2025 Jul 3;11(1):304. doi: 10.1038/s41420-025-02575-w. Cell Death Discov. 2025. PMID: 40610427 Free PMC article.
-
Mitochondria as a Disease-Relevant Organelle in Rheumatoid Arthritis: A Key Breakout in Fight Against the Disease.Biomedicines. 2025 Jul 13;13(7):1708. doi: 10.3390/biomedicines13071708. Biomedicines. 2025. PMID: 40722779 Free PMC article. Review.
References
-
- Abdallah Y, Kasseckert SA, Iraqi W, Said M, Shahzad T, Erdogan A, Neuhof C, Gündüz D, Schlüter K-D, Tillmanns H, Piper HM, Reusch HP, Ladilov Y (2011) Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med 15:2478–2485. 10.1111/j.1582-4934.2010.01249.x 10.1111/j.1582-4934.2010.01249.x - DOI - PMC - PubMed
-
- Antoniel M, Jones K, Antonucci S, Spolaore B, Fogolari F, Petronilli V, Giorgio V, Carraro M, Di Lisa F, Forte M, Szabó I, Lippe G, Bernardi P (2018) The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep 19:257–268. 10.15252/embr.201744705 10.15252/embr.201744705 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
