

Sepsis endotypes identified by host gene expression across global cohorts
- PMID: 38890515
- PMCID: PMC11189468
- DOI: 10.1038/s43856-024-00542-7
Sepsis endotypes identified by host gene expression across global cohorts
Abstract
Background: Sepsis from infection is a global health priority and clinical trials have failed to deliver effective therapeutic interventions. To address complicating heterogeneity in sepsis pathobiology, and improve outcomes, promising precision medicine approaches are helping identify disease endotypes, however, they require a more complete definition of sepsis subgroups.
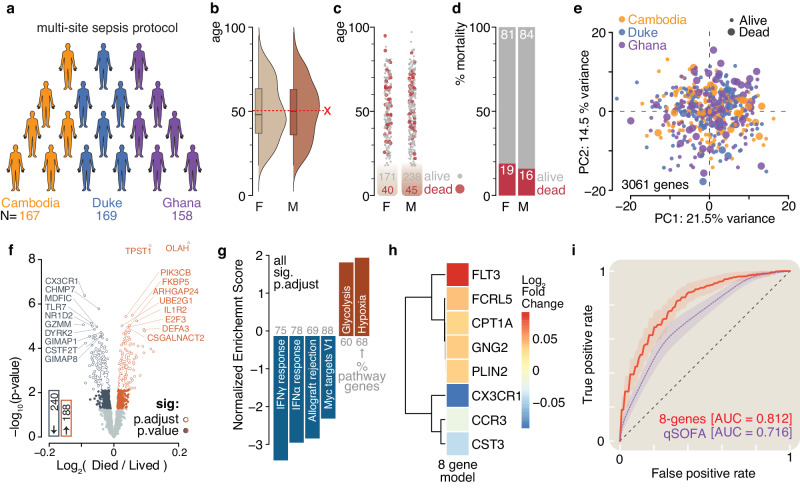
Methods: Here, we use RNA sequencing from peripheral blood to interrogate the host response to sepsis from participants in a global observational study carried out in West Africa, Southeast Asia, and North America (N = 494).
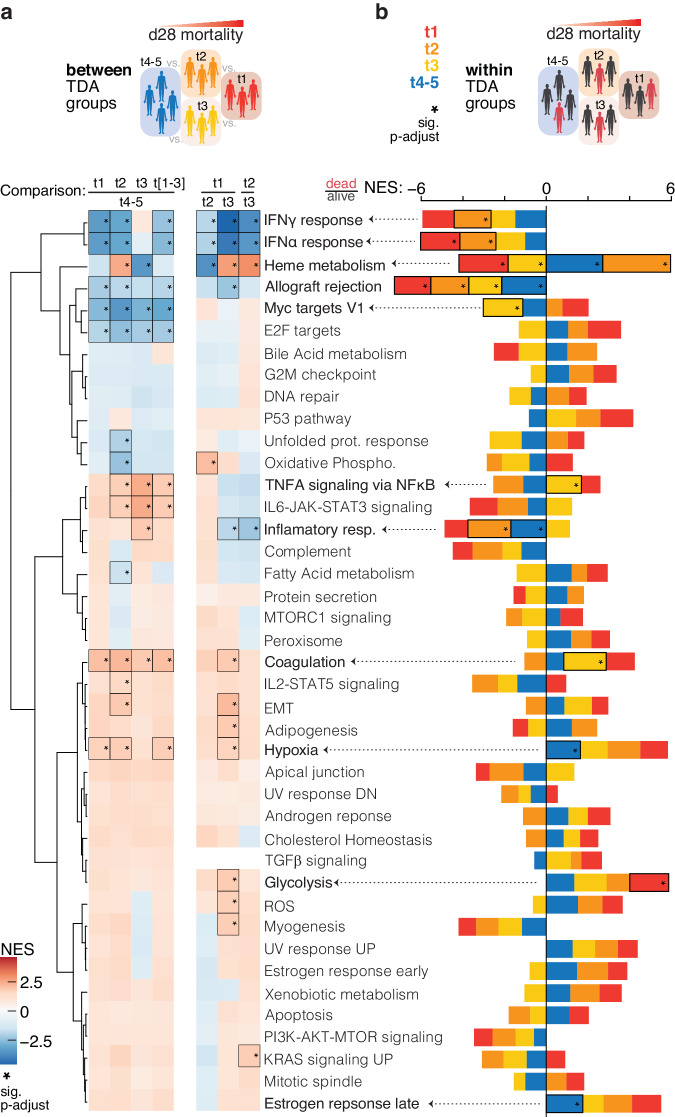
Results: We identify four sepsis subtypes differentiated by 28-day mortality. A low mortality immunocompetent group is specified by features that describe the adaptive immune system. In contrast, the three high mortality groups show elevated clinical severity consistent with multiple organ dysfunction. The immunosuppressed group members show signs of a dysfunctional immune response, the acute-inflammation group is set apart by molecular features of the innate immune response, while the immunometabolic group is characterized by metabolic pathways such as heme biosynthesis.
Conclusions: Our analysis reveals details of molecular endotypes in sepsis that support immunotherapeutic interventions and identifies biomarkers that predict outcomes in these groups.
Plain language summary
Sepsis is a life-threatening multi-organ failure caused by the body’s immune response to infection. Clinical symptoms of sepsis vary from one person to another likely due to differences in host factors, infecting pathogen, and comorbidities. This difference in clinical symptoms may contribute to the lack of effective interventions for sepsis. Therefore, approaches tailored to targeting groups of patients who present similarly are of great interest. This study analysed a large group of sepsis patients with diverse symptoms using laboratory markers and mathematical analysis. We report four patient groups that differ by risk of death and immune response profile. Targeting these defined groups with tailored interventions presents an exciting opportunity to improve the health outcomes of patients with sepsis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: D.V.C., J.B., J.G.C., and D.A.S. are listed as inventors on a U.S. Provisional Application No. 63/578,492, entitled: “Prediction of Mortality of Patients Diagnosed with Sepsis,” applied for by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., on the identification of molecular endotypes that predict sepsis mortality. E.L.T is an employee and has equity in Danaher. E.L.T and C.W.W have received consulting fees from Biomeme. All other authors have no competing interests to declare.
Figures




References
Grants and funding
LinkOut - more resources
Full Text Sources
