

Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities
- PMID: 38891760
- PMCID: PMC11171558
- DOI: 10.3390/ijms25115572
Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities
Abstract
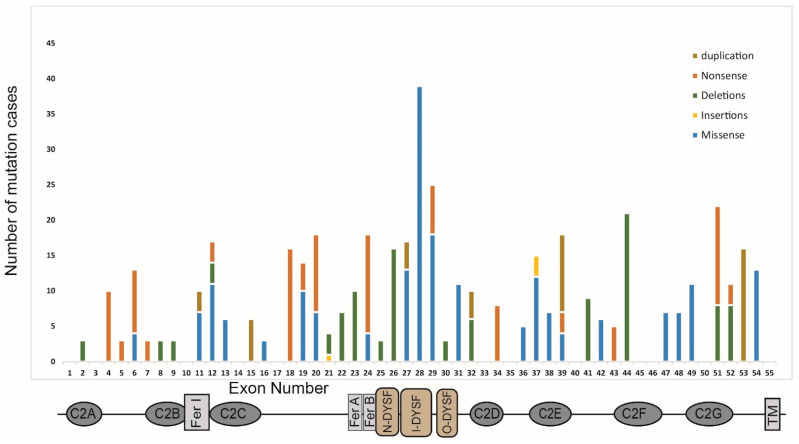
Dysferlin is a large transmembrane protein involved in critical cellular processes including membrane repair and vesicle fusion. Mutations in the dysferlin gene (DYSF) can result in rare forms of muscular dystrophy; Miyoshi myopathy; limb girdle muscular dystrophy type 2B (LGMD2B); and distal myopathy. These conditions are collectively known as dysferlinopathies and are caused by more than 600 mutations that have been identified across the DYSF gene to date. In this review, we discuss the key molecular and clinical features of LGMD2B, the causative gene DYSF, and the associated dysferlin protein structure. We also provide an update on current approaches to LGMD2B diagnosis and advances in drug development, including splice switching antisense oligonucleotides. We give a brief update on clinical trials involving adeno-associated viral gene therapy and the current progress on CRISPR/Cas9 mediated therapy for LGMD2B, and then conclude by discussing the prospects of antisense oligomer-based intervention to treat selected mutations causing dysferlinopathies.
Keywords: LGMD2B; antisense oligonucleotides; dysferlinopathies; gene therapy; readthrough therapy.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
