

Revisiting Glutamate Excitotoxicity in Amyotrophic Lateral Sclerosis and Age-Related Neurodegeneration
- PMID: 38891774
- PMCID: PMC11171854
- DOI: 10.3390/ijms25115587
Revisiting Glutamate Excitotoxicity in Amyotrophic Lateral Sclerosis and Age-Related Neurodegeneration
Abstract
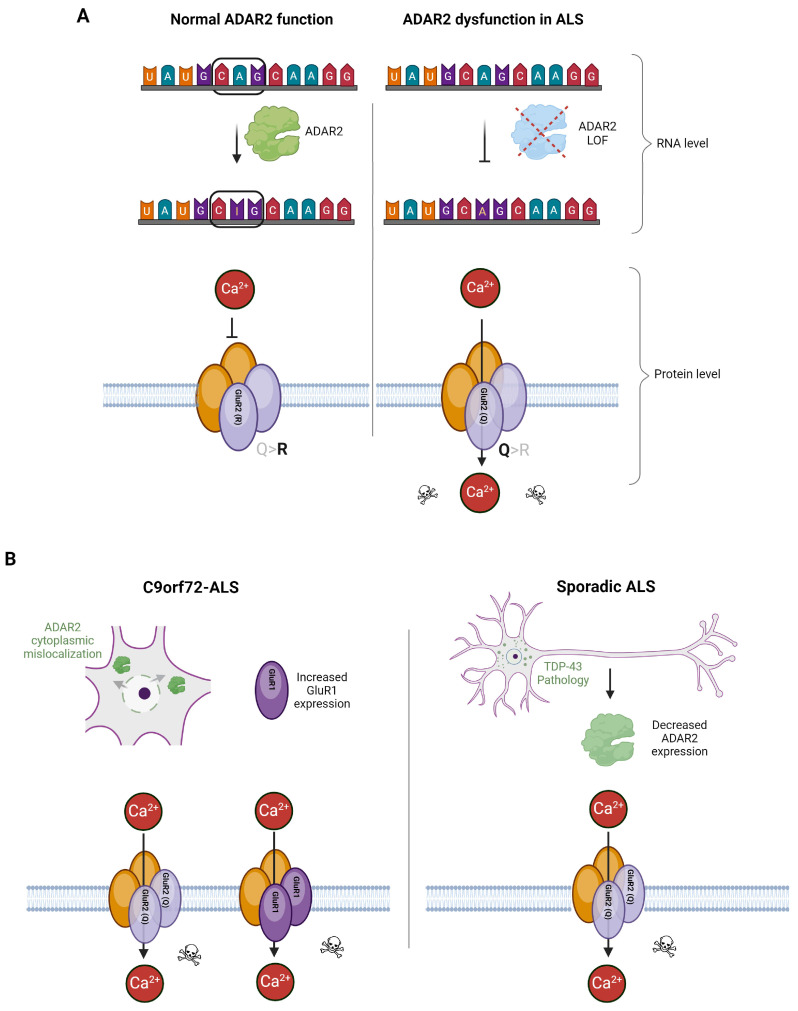
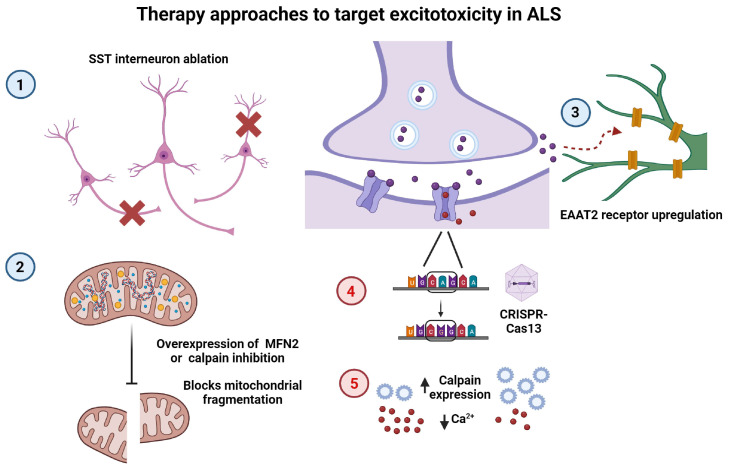
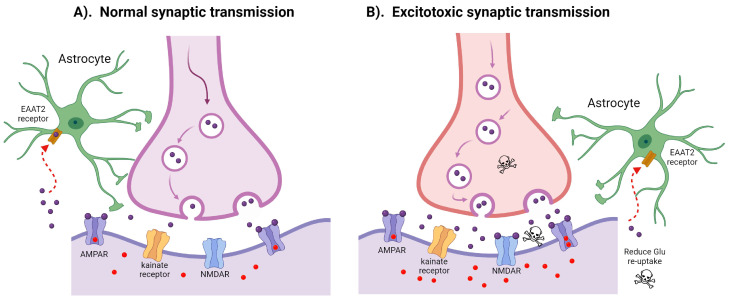
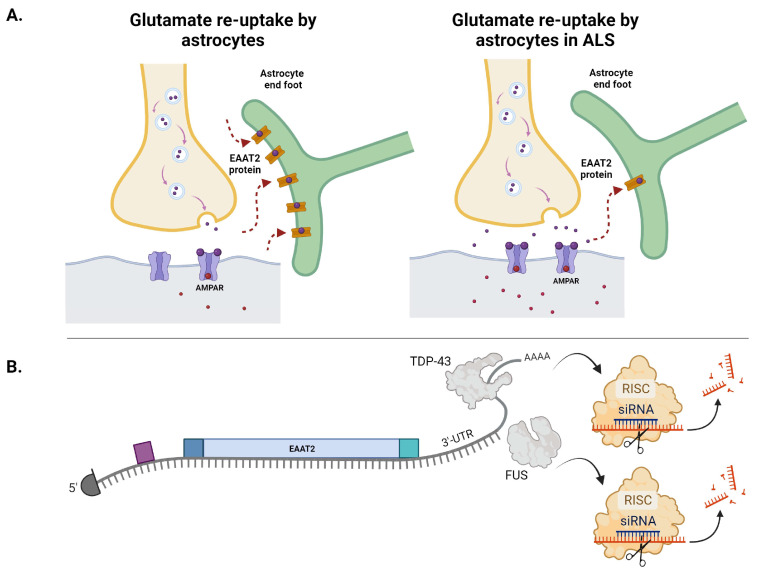
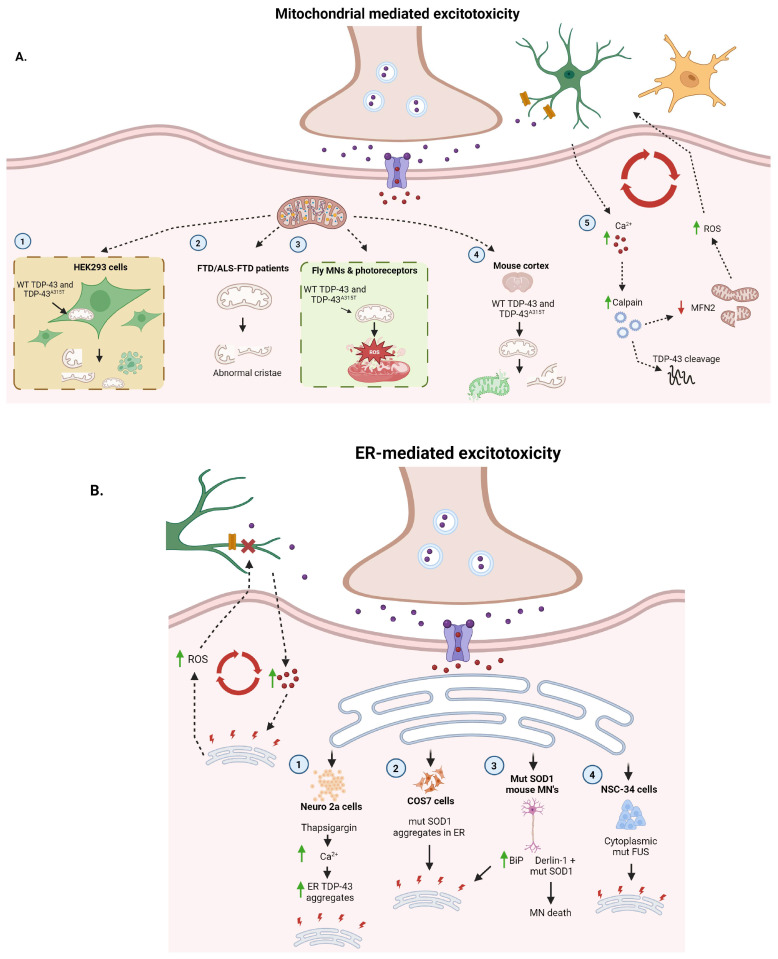
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disorder. While there are five FDA-approved drugs for treating this disease, each has only modest benefits. To design new and more effective therapies for ALS, particularly for sporadic ALS of unknown and diverse etiologies, we must identify key, convergent mechanisms of disease pathogenesis. This review focuses on the origin and effects of glutamate-mediated excitotoxicity in ALS (the cortical hyperexcitability hypothesis), in which increased glutamatergic signaling causes motor neurons to become hyperexcitable and eventually die. We characterize both primary and secondary contributions to excitotoxicity, referring to processes taking place at the synapse and within the cell, respectively. 'Primary pathways' include upregulation of calcium-permeable AMPA receptors, dysfunction of the EAAT2 astrocytic glutamate transporter, increased release of glutamate from the presynaptic terminal, and reduced inhibition by cortical interneurons-all of which have been observed in ALS patients and model systems. 'Secondary pathways' include changes to mitochondrial morphology and function, increased production of reactive oxygen species, and endoplasmic reticulum (ER) stress. By identifying key targets in the excitotoxicity cascade, we emphasize the importance of this pathway in the pathogenesis of ALS and suggest that intervening in this pathway could be effective for developing therapies for this disease.
Keywords: AMPA receptors; GluR2 editing; NMDA receptors; astrocytes; glutamate excitotoxicity.
Conflict of interest statement
We declare no financial, personal, or professional conflicts of interest relating to this manuscript/subject matter.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
