

Identification of Marker Genes in Infectious Diseases from ScRNA-seq Data Using Interpretable Machine Learning
- PMID: 38892107
- PMCID: PMC11172967
- DOI: 10.3390/ijms25115920
Identification of Marker Genes in Infectious Diseases from ScRNA-seq Data Using Interpretable Machine Learning
Abstract
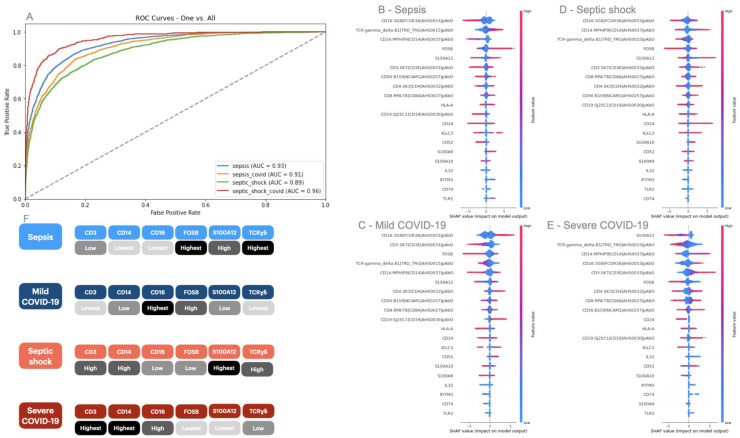
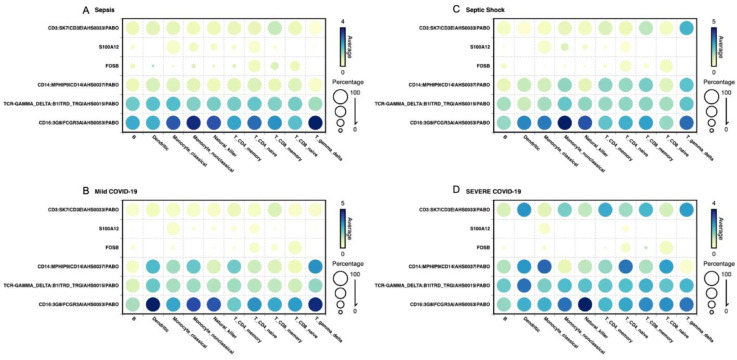
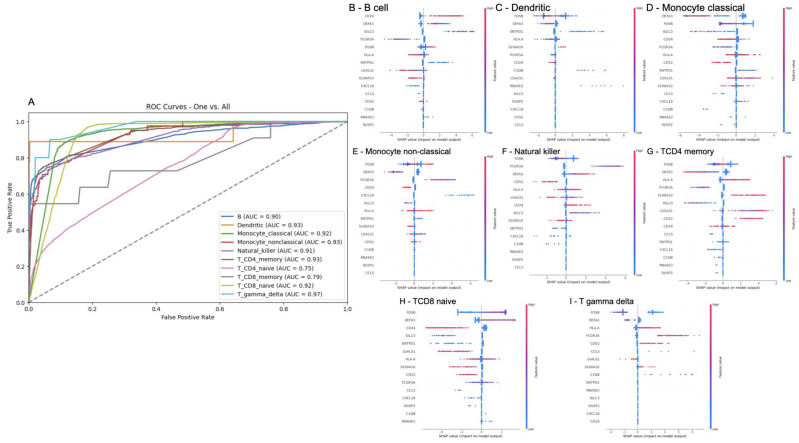
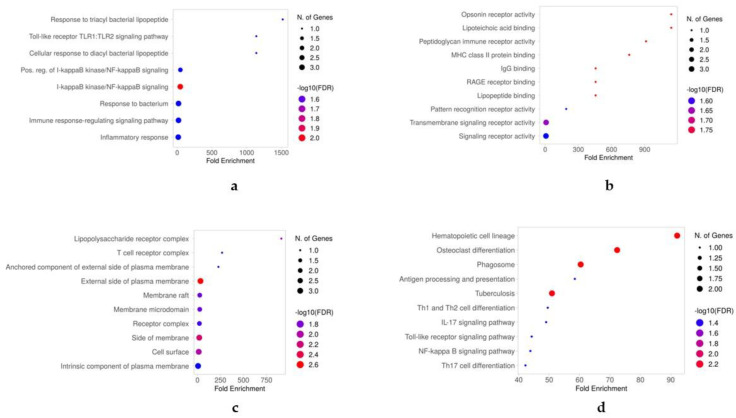
A common result of infection is an abnormal immune response, which may be detrimental to the host. To control the infection, the immune system might undergo regulation, therefore producing an excess of either pro-inflammatory or anti-inflammatory pathways that can lead to widespread inflammation, tissue damage, and organ failure. A dysregulated immune response can manifest as changes in differentiated immune cell populations and concentrations of circulating biomarkers. To propose an early diagnostic system that enables differentiation and identifies the severity of immune-dysregulated syndromes, we built an artificial intelligence tool that uses input data from single-cell RNA sequencing. In our results, single-cell transcriptomics successfully distinguished between mild and severe sepsis and COVID-19 infections. Moreover, by interpreting the decision patterns of our classification system, we identified that different immune cells upregulating or downregulating the expression of the genes CD3, CD14, CD16, FOSB, S100A12, and TCRɣδ can accurately differentiate between different degrees of infection. Our research has identified genes of significance that effectively distinguish between infections, offering promising prospects as diagnostic markers and providing potential targets for therapeutic intervention.
Keywords: artificial intelligence; marker genes; sepsis; single-cell RNA sequencing.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Exploring the Role of Circadian Rhythm-Related Genes in the Identification of Sepsis Subtypes and the Construction of Diagnostic Models Based on RNA-seq and scRNA-seq.Int J Mol Sci. 2025 Apr 23;26(9):3993. doi: 10.3390/ijms26093993. Int J Mol Sci. 2025. PMID: 40362233 Free PMC article.
-
scPanel: a tool for automatic identification of sparse gene panels for generalizable patient classification using scRNA-seq datasets.Brief Bioinform. 2024 Sep 23;25(6):bbae482. doi: 10.1093/bib/bbae482. Brief Bioinform. 2024. PMID: 39350339 Free PMC article.
-
Deciphering Abnormal Platelet Subpopulations in COVID-19, Sepsis and Systemic Lupus Erythematosus through Machine Learning and Single-Cell Transcriptomics.Int J Mol Sci. 2024 May 29;25(11):5941. doi: 10.3390/ijms25115941. Int J Mol Sci. 2024. PMID: 38892129 Free PMC article.
-
Biological and Medical Importance of Cellular Heterogeneity Deciphered by Single-Cell RNA Sequencing.Cells. 2020 Jul 22;9(8):1751. doi: 10.3390/cells9081751. Cells. 2020. PMID: 32707839 Free PMC article. Review.
-
Machine learning and statistical methods for clustering single-cell RNA-sequencing data.Brief Bioinform. 2020 Jul 15;21(4):1209-1223. doi: 10.1093/bib/bbz063. Brief Bioinform. 2020. PMID: 31243426 Review.
Cited by
-
MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity.Int J Mol Sci. 2025 May 15;26(10):4743. doi: 10.3390/ijms26104743. Int J Mol Sci. 2025. PMID: 40429886 Free PMC article.
-
Single-cell expression and immune infiltration analysis of polyamine metabolism in breast cancer.Discov Oncol. 2024 Nov 16;15(1):666. doi: 10.1007/s12672-024-01524-w. Discov Oncol. 2024. PMID: 39549127 Free PMC article.
References
-
- Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G.R., Chiche J.-D., Coopersmith C.M., et al. The third international consensus definitions for sepsis and septic shock (sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. - DOI - PMC - PubMed
-
- Rudd K.E., Johnson S.C., Agesa K.M., Shackelford K.A., Tsoi D., Kievlan D.R., Colombara D.V., Ikuta K.S., Kissoon N., Finfer S., et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet. 2020;395:200–211. doi: 10.1016/s0140-6736(19)32989-7. - DOI - PMC - PubMed
-
- Bermejo-Martin J.F., Gonzalez-Rivera M., Almansa R., Micheloud D., Tedim A.P., Dominguez-Gil M., Resino S., Martin-Fernandez M., Murua P.R., Perez-Garcia F., et al. Viral RNA load in plasma is associated with critical illness and a dysregulated host response in COVID-19. Crit. Care. 2020;24:691. doi: 10.1186/s13054-020-03398-0. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
