

Endothelial-to-Mesenchymal Transition in Cardiovascular Pathophysiology
- PMID: 38892367
- PMCID: PMC11173124
- DOI: 10.3390/ijms25116180
Endothelial-to-Mesenchymal Transition in Cardiovascular Pathophysiology
Abstract
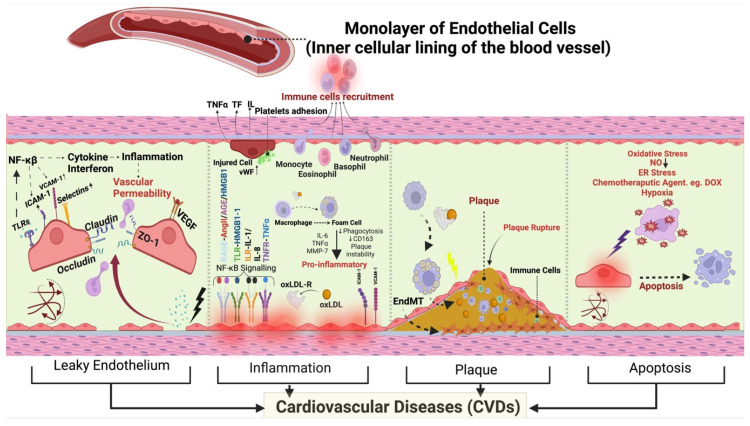
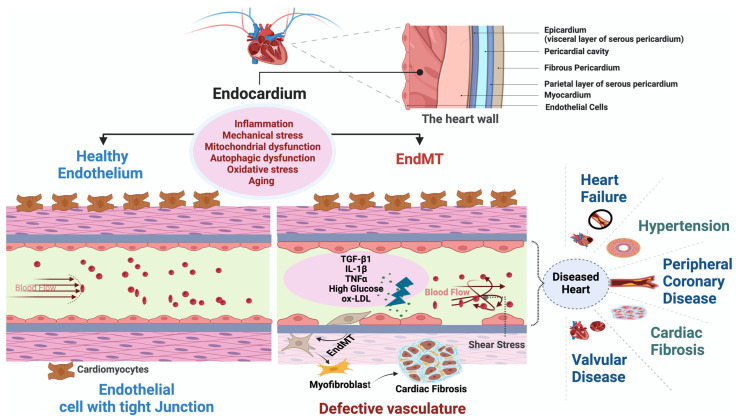
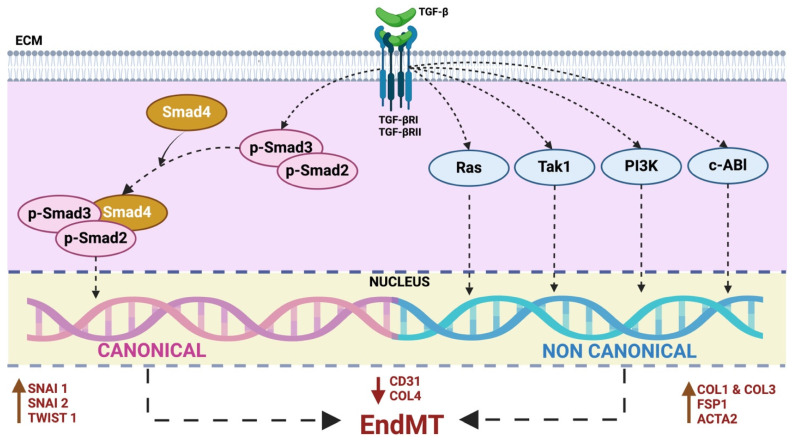
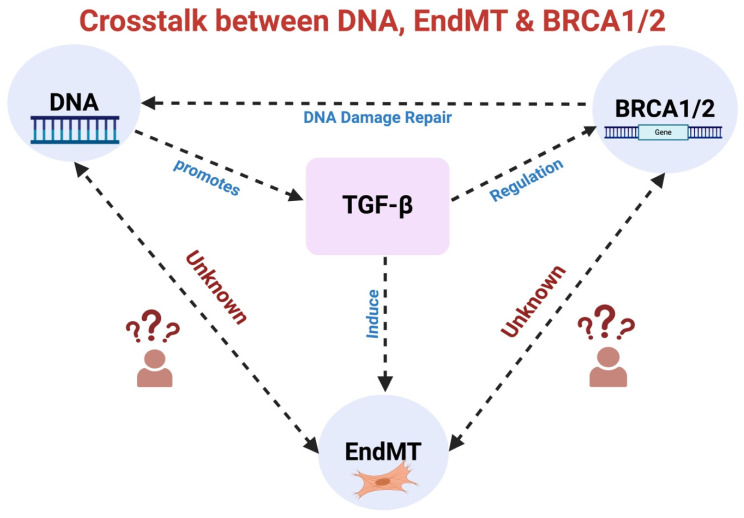
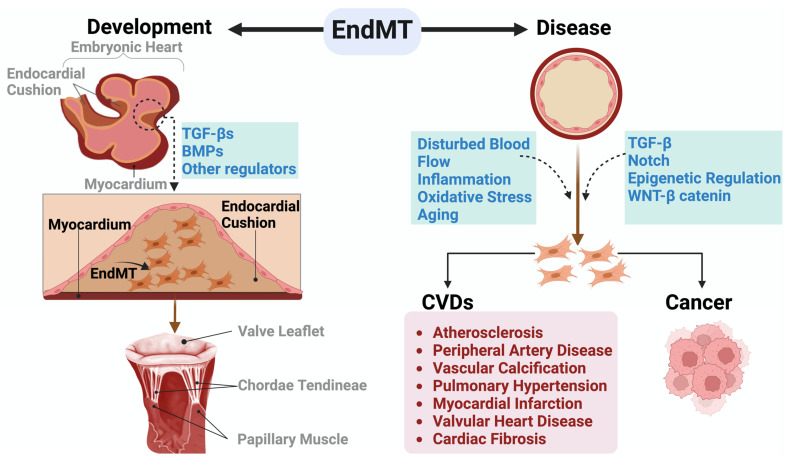
Under different pathophysiological conditions, endothelial cells lose endothelial phenotype and gain mesenchymal cell-like phenotype via a process known as endothelial-to-mesenchymal transition (EndMT). At the molecular level, endothelial cells lose the expression of endothelial cell-specific markers such as CD31/platelet-endothelial cell adhesion molecule, von Willebrand factor, and vascular-endothelial cadherin and gain the expression of mesenchymal cell markers such as α-smooth muscle actin, N-cadherin, vimentin, fibroblast specific protein-1, and collagens. EndMT is induced by numerous different pathways triggered and modulated by multiple different and often redundant mechanisms in a context-dependent manner depending on the pathophysiological status of the cell. EndMT plays an essential role in embryonic development, particularly in atrioventricular valve development; however, EndMT is also implicated in the pathogenesis of several genetically determined and acquired diseases, including malignant, cardiovascular, inflammatory, and fibrotic disorders. Among cardiovascular diseases, aberrant EndMT is reported in atherosclerosis, pulmonary hypertension, valvular disease, fibroelastosis, and cardiac fibrosis. Accordingly, understanding the mechanisms behind the cause and/or effect of EndMT to eventually target EndMT appears to be a promising strategy for treating aberrant EndMT-associated diseases. However, this approach is limited by a lack of precise functional and molecular pathways, causes and/or effects, and a lack of robust animal models and human data about EndMT in different diseases. Here, we review different mechanisms in EndMT and the role of EndMT in various cardiovascular diseases.
Keywords: EndMT; cardiovascular diseases; endothelial-to-mesenchymal transition; mechanisms.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
