

NOTCH3 and Pulmonary Arterial Hypertension
- PMID: 38892440
- PMCID: PMC11172835
- DOI: 10.3390/ijms25116248
NOTCH3 and Pulmonary Arterial Hypertension
Abstract
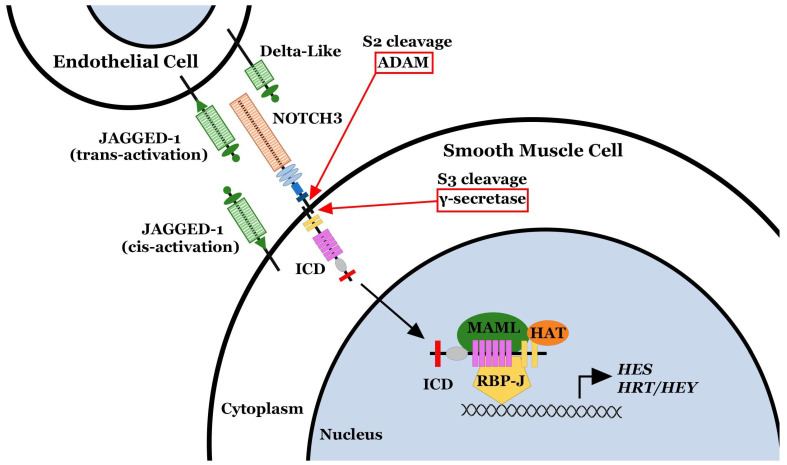
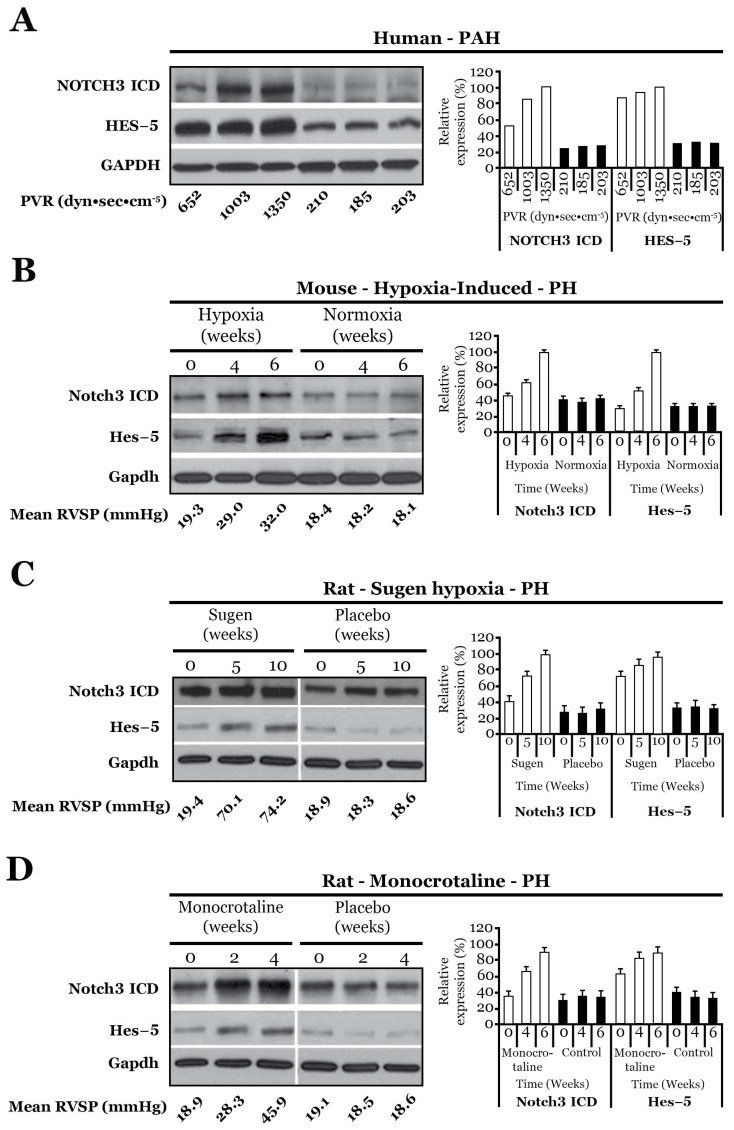
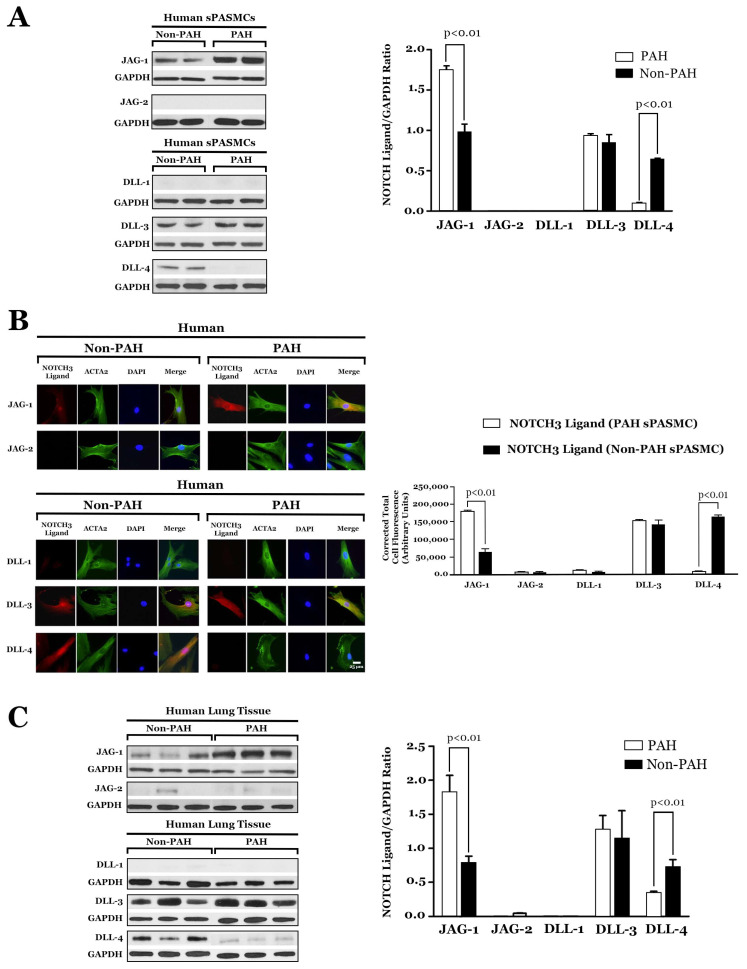
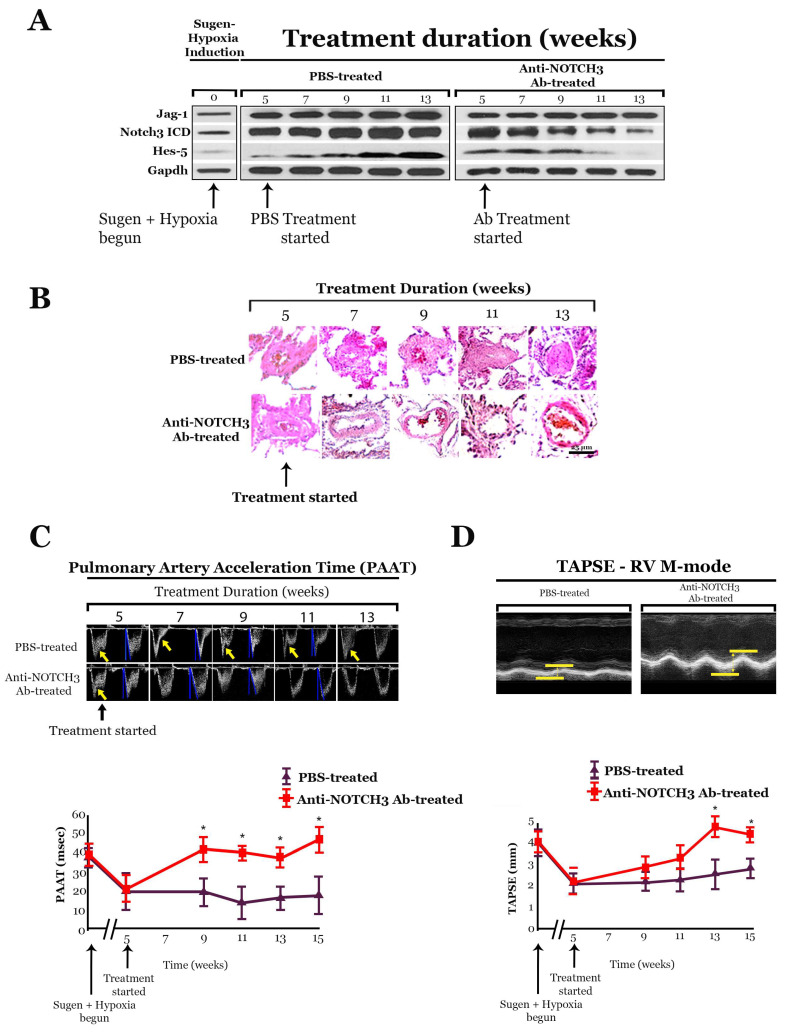
NOTCH3 receptor signaling has been linked to the regulation of smooth muscle cell proliferation and the maintenance of smooth muscle cells in an undifferentiated state. Pulmonary arterial hypertension (World Health Organization Group 1 idiopathic disease: PAH) is a fatal disease characterized clinically by elevated pulmonary vascular resistance caused by extensive vascular smooth muscle cell proliferation, perivascular inflammation, and asymmetric neointimal hyperplasia in precapillary pulmonary arteries. In this review, a detailed overview of the specific role of NOTCH3 signaling in PAH, including its mechanisms of activation by a select ligand, downstream signaling effectors, and physiologic effects within the pulmonary vascular tree, is provided. Animal models showing the importance of the NOTCH3 pathway in clinical PAH will be discussed. New drugs and biologics that inhibit NOTCH3 signaling and reverse this deadly disease are highlighted.
Keywords: DLL-4; HES-5; JAG-1; NOTCH3; Notch3; PAH; pulmonary arterial hypertension.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
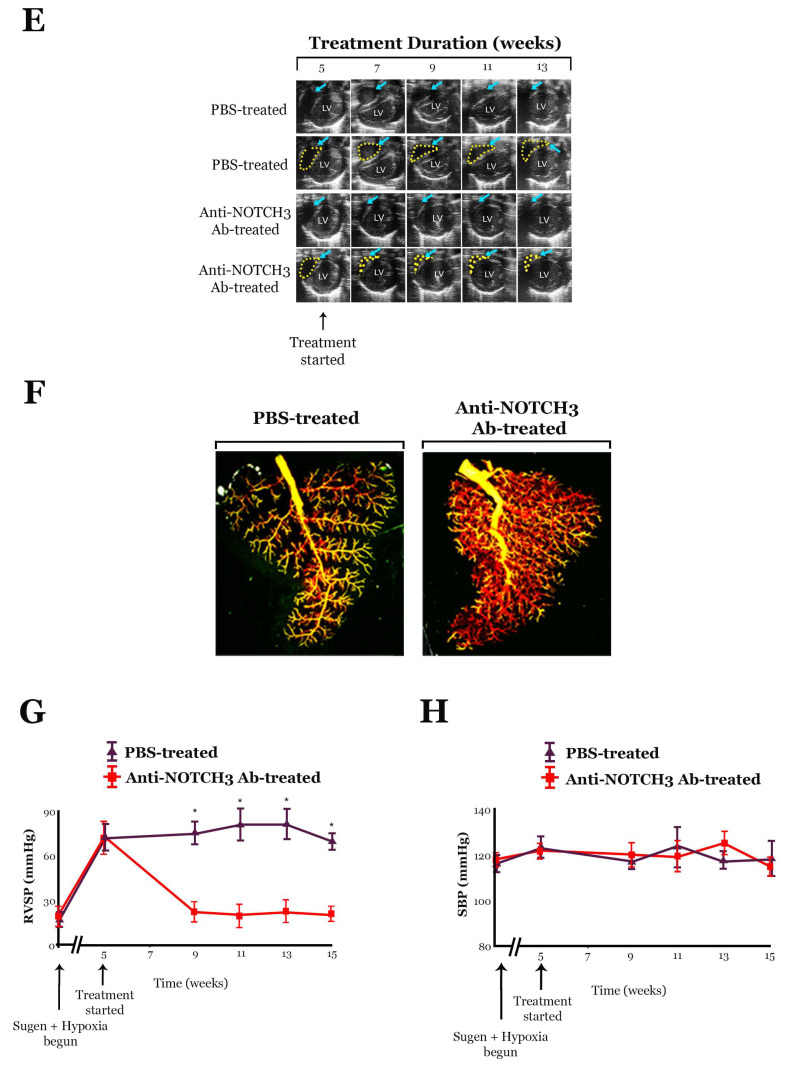
References
-
- Badagliacca R., Poscia R., Pezzuto B., Nocioni M., Mezzapesa M., Francone M., Giannetta E., Papa S., Gambardella C., Sciomer S., et al. Right ventricular remodeling in idiopathic pulmonary arterial hypertension: Adaptive versus maladaptive morphology. J. Heart Lung Transplant. 2015;34:395–403. doi: 10.1016/j.healun.2014.11.002. - DOI - PubMed
-
- Hardegree E.L., Sachdev A., Fenstad E.R., Villarraga H.R., Frantz R.P., McGoon M.D., Oh J.K., Ammash N.M., Connolly H.M., Eidem B.W., et al. Impaired left ventricular mechanics in pulmonary arterial hypertension: Identification of a cohort at high risk. Circ. Heart Fail. 2013;6:748–755. doi: 10.1161/circheartfailure.112.000098. - DOI - PubMed
-
- Steffes L.C., Froistad A.A., Andruska A., Boehm M., McGlynn M., Zhang F., Zhang W., Hou D., Tian X., Miquerol L., et al. A Notch3-Marked Subpopulation of Vascular Smooth Muscle Cells Is the Cell of Origin for Occlusive Pulmonary Vascular Lesions. Circulation. 2020;142:1545–1561. doi: 10.1161/circulationaha.120.045750. - DOI - PMC - PubMed
-
- Humbert M., Guignabert C., Bonnet S., Dorfmüller P., Klinger J.R., Nicolls M.R., Olschewski A.J., Pullamsetti S.S., Schermuly R.T., Stenmark K.R., et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019;53:1801887. doi: 10.1183/13993003.01887-2018. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
