

Hereditary Syndromes Associated with Pancreatic and Lung Neuroendocrine Tumors
- PMID: 38893191
- PMCID: PMC11171219
- DOI: 10.3390/cancers16112075
Hereditary Syndromes Associated with Pancreatic and Lung Neuroendocrine Tumors
Abstract
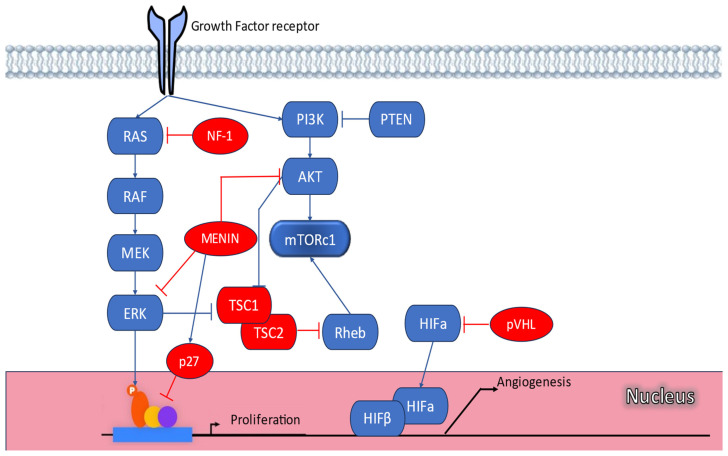
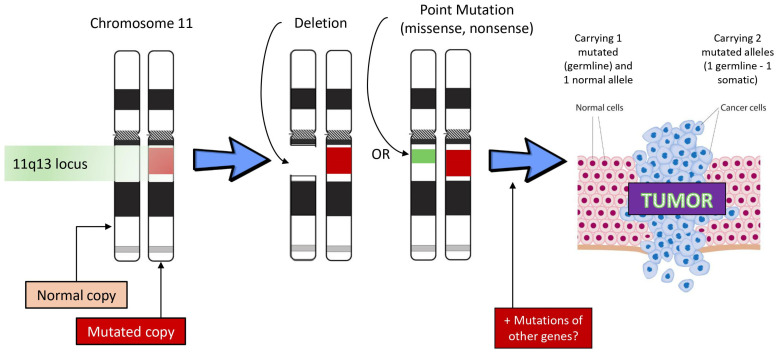
Pancreatic neuroendocrine tumors (PanNETs) and lung NETs (LNETs) represent a rare but clinically significant subgroup of neoplasms. While the majority is sporadic, approximately 17% of PanNETs and a subset of LNETs develop in the context of monogenic familial tumor syndromes, especially multiple endocrine neoplasia type 1 (MEN1) syndrome. Other inherited syndromes associated with PanNETs include MEN4, von Hippel-Lindau (VHL) syndrome, neurofibromatosis type 1 (NF1), and tuberous sclerosis complex (TSC). These syndromes are highly penetrant and their clinical manifestations may vary even among members of the same family. They are attributed to genetic mutations involving key molecular pathways regulating cell growth, differentiation, and angiogenesis. Pancreatic NETs in hereditary syndromes are often multiple, develop at a younger age compared to sporadic tumors, and are associated with endocrine and nonendocrine tumors derived from multiple organs. Lung NETs are not as common as PanNETs and are mostly encountered in MEN1 syndrome and include typical and atypical lung carcinoids. Early detection of PanNETs and LNETs related to inherited syndromes is crucial, and specific follow-up protocols need to be employed to optimize diagnosis and management. Genetic screening is recommended in childhood, and diagnostic screening starts often in adolescence, even in asymptomatic mutation carriers. Optimal management and therapeutic decisions should be made in the context of a multidisciplinary team in specialized centers, whereas specific biomarkers aiming to identify patients denoted to follow a more aggressive course need to be developed.
Keywords: MEN1; hereditary syndromes; lung neuroendocrine tumors; pancreatic neuroendocrine tumors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
