

Hydration of N-Hydroxyurea from Ab Initio Molecular Dynamics Simulations
- PMID: 38893311
- PMCID: PMC11173572
- DOI: 10.3390/molecules29112435
Hydration of N-Hydroxyurea from Ab Initio Molecular Dynamics Simulations
Abstract
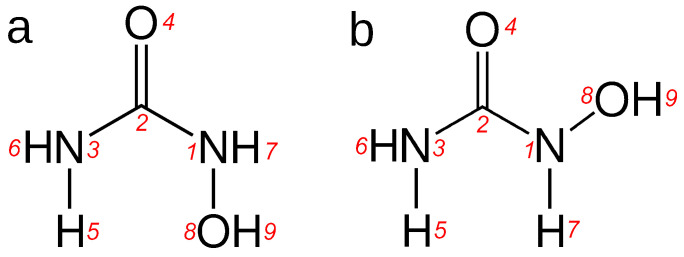
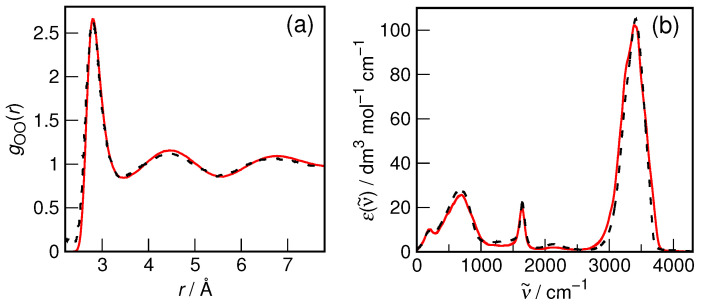
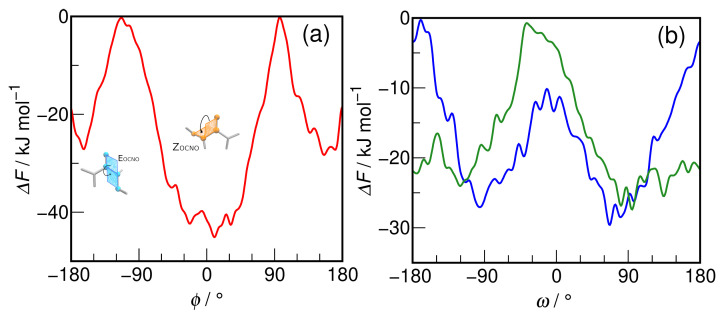
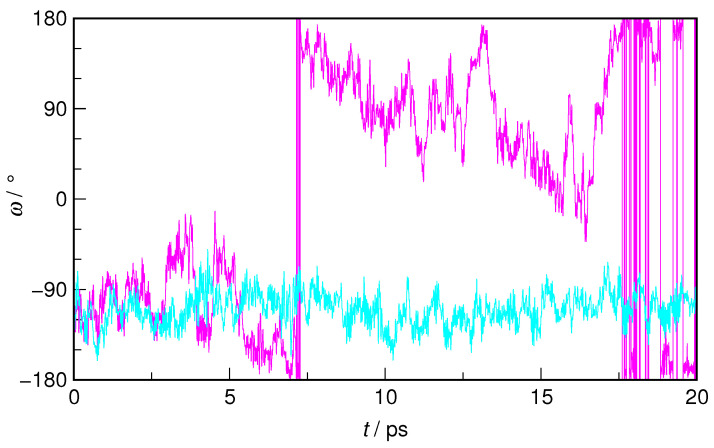
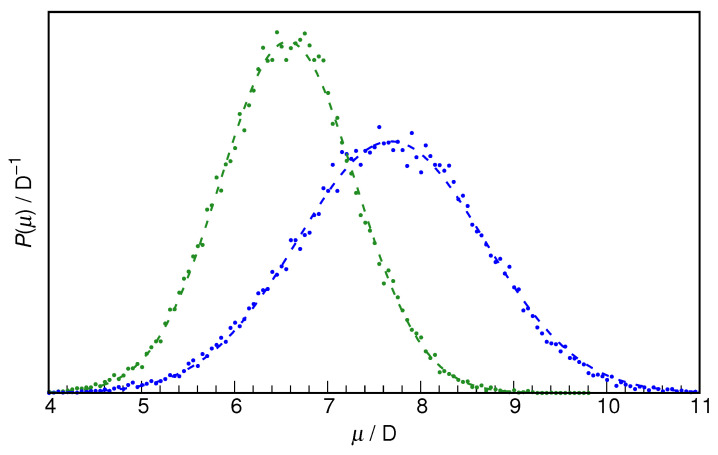
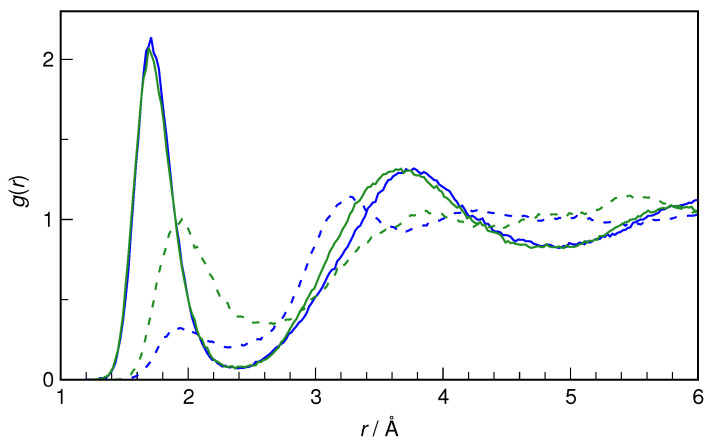
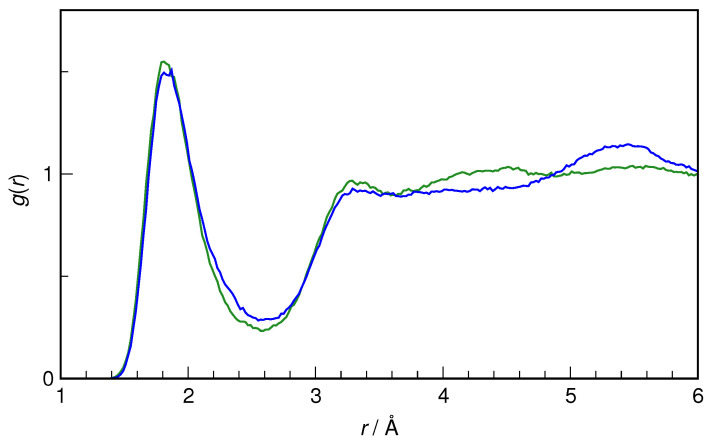
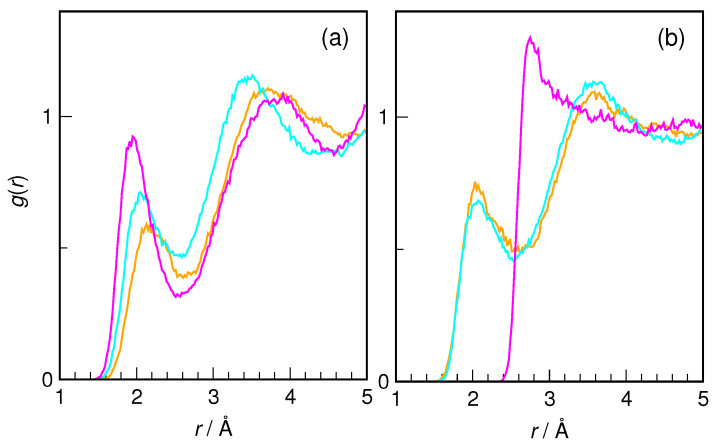
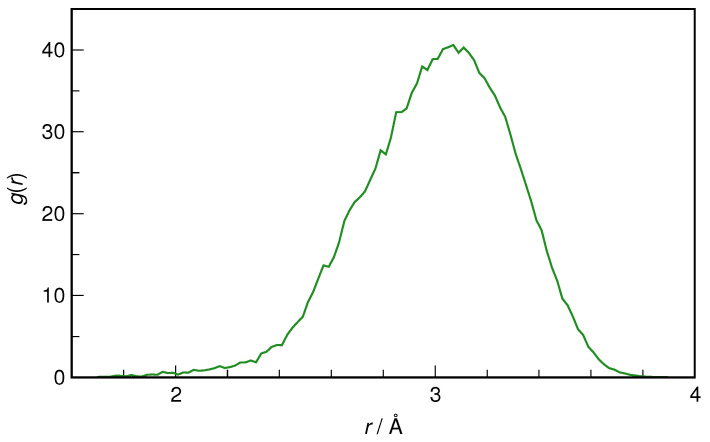
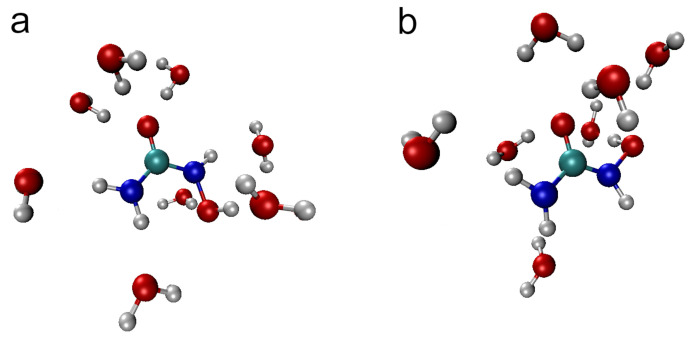
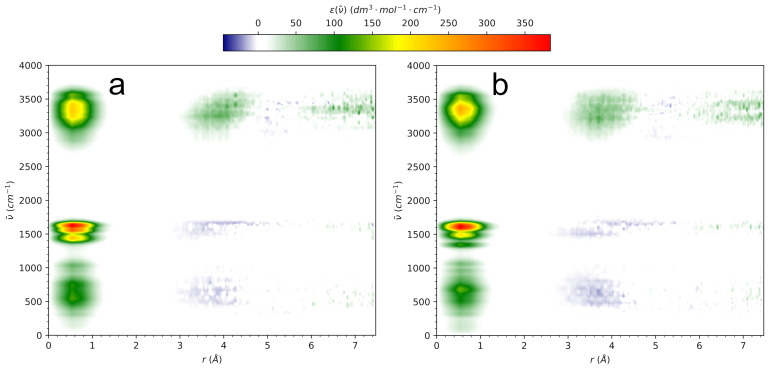
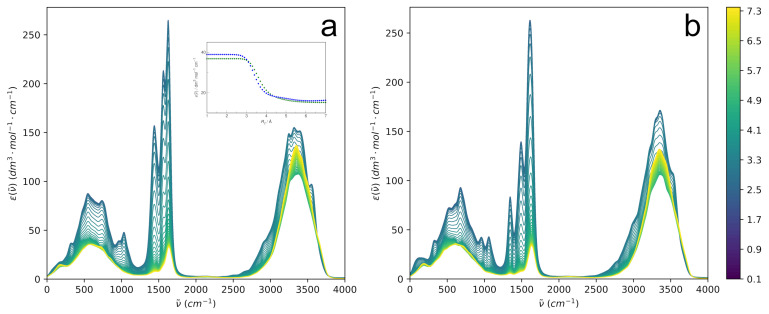
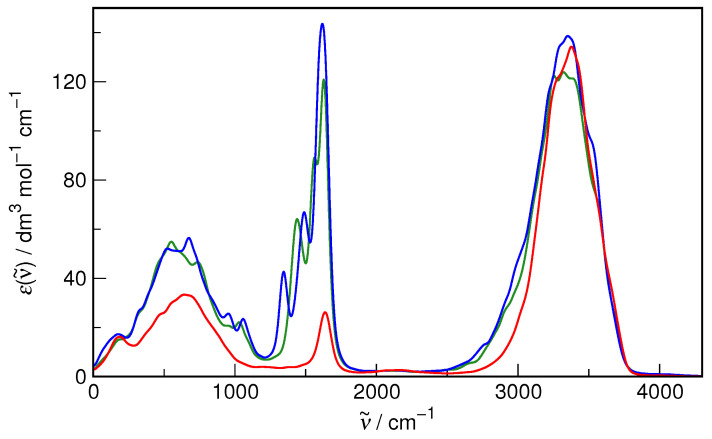
N-Hydroxyurea (HU) is an important chemotherapeutic agent used as a first-line treatment in conditions such as sickle cell disease and β-thalassemia, among others. To date, its properties as a hydrated molecule in the blood plasma or cytoplasm are dramatically understudied, although they may be crucial to the binding of HU to the radical catalytic site of ribonucleotide reductase, its molecular target. The purpose of this work is the comprehensive exploration of HU hydration. The topic is studied using ab initio molecular dynamic (AIMD) simulations that apply a first principles representation of the electron density of the system. This allows for the calculation of infrared spectra, which may be decomposed spatially to better capture the spectral signatures of solute-solvent interactions. The studied molecule is found to be strongly hydrated and tightly bound to the first shell water molecules. The analysis of the distance-dependent spectra of HU shows that the E and Z conformers spectrally affect, on average, 3.4 and 2.5 of the closest H2O molecules, respectively, in spheres of radii of 3.7 Å and 3.5 Å, respectively. The distance-dependent spectra corresponding to these cutoff radii show increased absorbance in the red-shifted part of the water OH stretching vibration band, indicating local enhancement of the solvent's hydrogen bond network. The radially resolved IR spectra also demonstrate that HU effortlessly incorporates into the hydrogen bond network of water and has an enhancing effect on this network. Metadynamics simulations based on AIMD methodology provide a picture of the conformational equilibria of HU in solution. Contrary to previous investigations of an isolated HU molecule in the gas phase, the Z conformer of HU is found here to be more stable by 17.4 kJ·mol-1 than the E conformer, pointing at the crucial role that hydration plays in determining the conformational stability of solutes. The potential energy surface for the OH group rotation in HU indicates that there is no intramolecular hydrogen bond in Z-HU in water, in stark contrast to the isolated solute in the gas phase. Instead, the preferred orientation of the hydroxyl group is perpendicular to the molecular plane of the solute. In view of the known chaotropic effect of urea and its N-alkyl-substituted derivatives, N-hydroxyurea emerges as a unique urea derivative that exhibits a kosmotropic ordering of nearby water. This property may be of crucial importance for its binding to the catalytic site of ribonucleotide reductase with a concomitant displacement of a water molecule.
Keywords: N-hydroxyurea; ab initio molecular dynamics; density functional theory; hydration.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Structure and dynamics of the hydration shells of the Zn(2+) ion from ab initio molecular dynamics and combined ab initio and classical molecular dynamics simulations.J Chem Phys. 2010 May 21;132(19):194502. doi: 10.1063/1.3421542. J Chem Phys. 2010. PMID: 20499974
-
Communication: Inside the water wheel: Intrinsic differences between hydrated tetraphenylphosphonium and tetraphenylborate ions.J Chem Phys. 2018 Nov 7;149(17):171101. doi: 10.1063/1.5056237. J Chem Phys. 2018. PMID: 30408992
-
The hydrogen bonding and hydration of 2'-OH in adenosine and adenosine 3'-ethyl phosphate.J Org Chem. 2002 Mar 22;67(6):1852-65. doi: 10.1021/jo010960j. J Org Chem. 2002. PMID: 11895403
-
The Clusters-in-a-Liquid Approach for Solvation: New Insights from the Conformer Specific Gas Phase Spectroscopy and Vibrational Optical Activity Spectroscopy.Front Chem. 2016 Feb 25;4:9. doi: 10.3389/fchem.2016.00009. eCollection 2016. Front Chem. 2016. PMID: 26942177 Free PMC article. Review.
-
Some ways of looking at compensatory kosmotropes and different water environments.Comp Biochem Physiol A Mol Integr Physiol. 2001 Oct;130(3):471-86. doi: 10.1016/s1095-6433(01)00416-0. Comp Biochem Physiol A Mol Integr Physiol. 2001. PMID: 11913459 Review.
References
LinkOut - more resources
Full Text Sources
