

Alpha globin gene alterations modifying the phenotype of homozygous beta thalassaemia
- PMID: 38895064
- PMCID: PMC11182400
- DOI: 10.1002/jha2.923
Alpha globin gene alterations modifying the phenotype of homozygous beta thalassaemia
Abstract
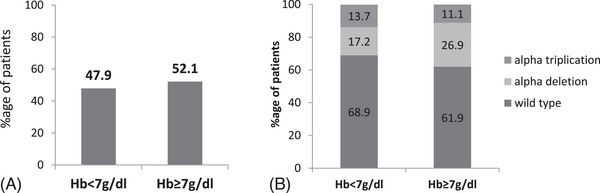
The phenotype of β-thalassemia varies widely. The primary determinant is the type of beta-globin gene mutation; however, there are secondary and tertiary modifiers also as associated alpha mutations, polymorphisms, as well as coinheritance of mutations affecting other related systems. Co-inheritance of alpha thalassemia mutations is known to ameliorate the severity of HbE-β thalassemia. However, the role of alpha globin gene alterations (deletions and triplication) is not well illustrated in homozygous β-thalassemia. Here we evaluated the role of alpha globin gene alterations in 122 β-thalassemia patients having IVS1-5 (G > C) homozygous mutation. β-thalassemia mutations were detected by ARMS PCR and alpha mutations by GAP-PCR. Gene expression by qRT-PCR. Out of 122 cases, 15 patients had alpha 3.7 triplications (ααα3.7anti), 24 had alpha 3.7 kb deletion (-α3.7) mutation and three patients had 4.2 kb deletion (-α4.2). Patients were divided into two groups, requiring less than 8 units (NTDT) and more than 8 units (TDT) of blood transfusion per year (≥8U BT/year). The percentage of alpha deletion was significantly (p = 0.0042) high in NTDT (42.1%) as compared with TDT (13.2%). Conversely, the proportion of alpha triplication is high in the TDT as compared with NTDT. Even mean serum ferritin level was found to be significantly high in patients having alpha triplication as compared with those having alpha deletions (p = 0.0184) and normal alpha gene (p = 0.0003). α/β globin ratio was highest in TDT patients with alpha triplication and lowest in NTDT patients with alpha-del. The results show that concurrent inheritance of alpha gene alterations influences the phenotypic severity of homozygous β-thalassemia.
Keywords: alpha deletion; alpha‐triplication; beta‐thalassemia; co‐inheritance; homozygous.
© 2024 The Author(s). eJHaem published by British Society for Haematology and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Balgir RS. The burden of haemoglobinopathies in India and the challenges ahead. Curr Sci. 2000:1536–1547.
-
- Colah R, Italia K, &Gorakshakar A Burden of thalassemia in India: the road map for control. Pediatr Hematol Oncol J. 2017;2(4):79–84. 10.1016/j.phoj.2017.10.002 - DOI
-
- Thaker P, Mahajan N, Mukherjee MB, Colah RB Molecular heterogeneity of Hb H disease in India. Thalassemia Reports. 2022;12(3):73–84. 10.3390/thalassrep12030012 - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous