

This is a preprint.
Predicting the direction of phenotypic difference
- PMID: 38895291
- PMCID: PMC11185551
- DOI: 10.1101/2024.02.22.581566
Predicting the direction of phenotypic difference
Update in
-
Predicting the direction of phenotypic difference.Nat Commun. 2025 Jul 26;16(1):6898. doi: 10.1038/s41467-025-62355-z. Nat Commun. 2025. PMID: 40715205 Free PMC article.
Abstract
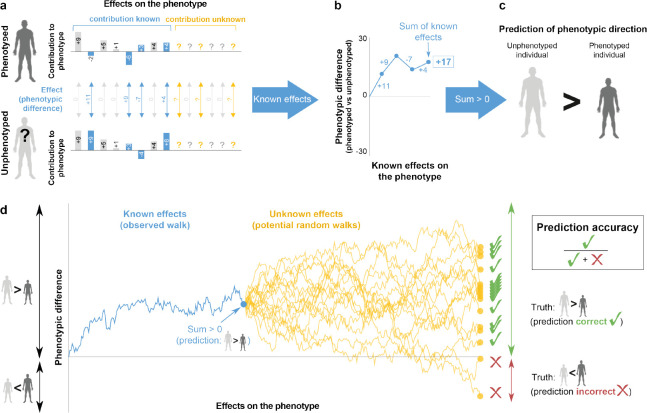
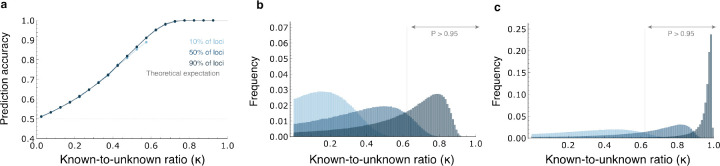
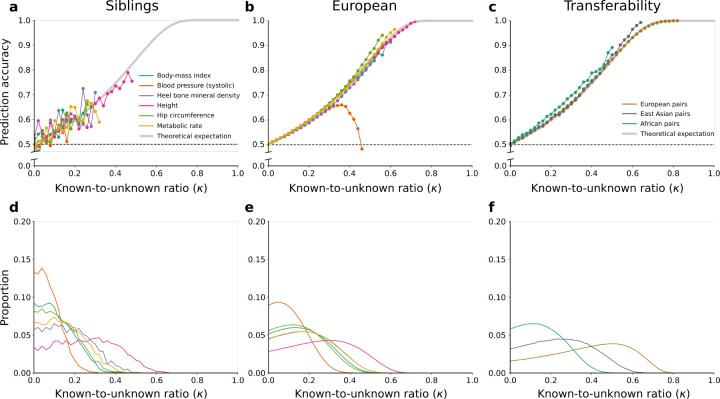
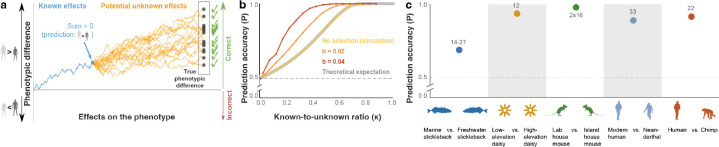
Predicting phenotypes from genomic data is a key goal in genetics, but for most complex phenotypes, predictions are hampered by incomplete genotype-to-phenotype mapping. Here, we describe a more attainable approach than quantitative predictions, which is aimed at qualitatively predicting phenotypic differences. Despite incomplete genotype-to-phenotype mapping, we show that it is relatively easy to determine which of two individuals has a greater phenotypic value. This question is central in many scenarios, e.g., comparing disease risk between individuals, the yield of crop strains, or the anatomy of extinct vs extant species. To evaluate prediction accuracy, i.e., the probability that the individual with the greater predicted phenotype indeed has a greater phenotypic value, we developed an estimator of the ratio between known and unknown effects on the phenotype. We evaluated prediction accuracy using human data from tens of thousands of individuals from either the same family or the same population, as well as data from different species. We found that, in many cases, even when only a small fraction of the loci affecting a phenotype is known, the individual with the greater phenotypic value can be identified with over 90% accuracy. Our approach also circumvents some of the limitations in transferring genetic association results across populations. Overall, we introduce an approach that enables accurate predictions of key information on phenotypes - the direction of phenotypic difference - and suggest that more phenotypic information can be extracted from genomic data than previously appreciated.
Figures
Similar articles
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Predicting the direction of phenotypic difference.Nat Commun. 2025 Jul 26;16(1):6898. doi: 10.1038/s41467-025-62355-z. Nat Commun. 2025. PMID: 40715205 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2020 Jan 9;1(1):CD011535. doi: 10.1002/14651858.CD011535.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2021 Apr 19;4:CD011535. doi: 10.1002/14651858.CD011535.pub4. PMID: 31917873 Free PMC article. Updated.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
References
-
- Orr H. A. The genetic theory of adaptation: a brief history. Nature Reviews Genetics 6, 119–127 (2005). - PubMed
-
- Scheben A. & Edwards D. Towards a more predictable plant breeding pipeline with CRISPR/Cas-induced allelic series to optimize quantitative and qualitative traits. Current Opinion in Plant Biology 45, 218–225 (2018). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources