

This is a preprint.
Protein Editing using a Concerted Transposition Reaction
- PMID: 38895383
- PMCID: PMC11185735
- DOI: 10.1101/2024.06.03.597171
Protein Editing using a Concerted Transposition Reaction
Update in
-
Protein editing using a coordinated transposition reaction.Science. 2025 Apr 4;388(6742):68-74. doi: 10.1126/science.adq8540. Epub 2025 Apr 3. Science. 2025. PMID: 40179182
Abstract
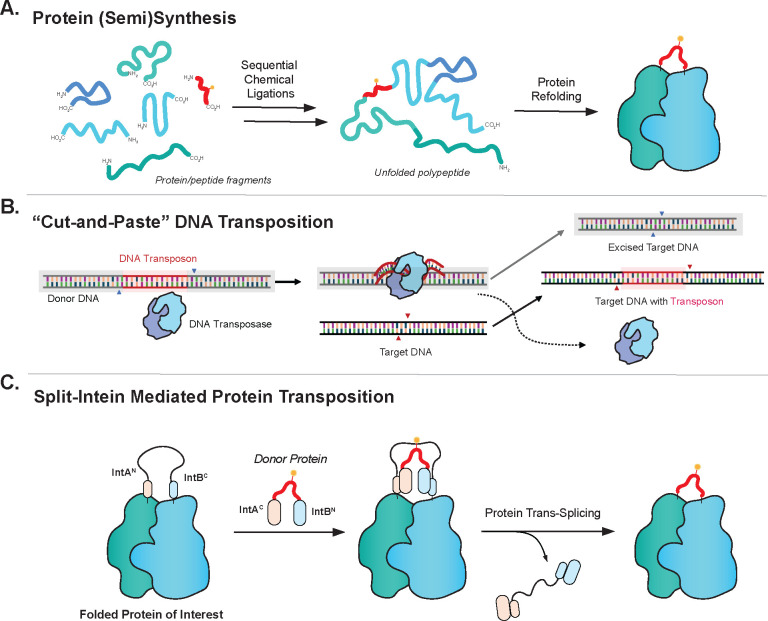
Protein engineering through the chemical or enzymatic ligation of polypeptide fragments has proven enormously powerful for studying countless biochemical processes in vitro. In general, this strategy necessitates a protein folding step following ligation of the unstructured fragments, a requirement that constrains the types of systems amenable to the approach. Here, we report an in vitro strategy that allows internal regions of target proteins to be replaced in a single operation. Conceptually, our system is analogous to a DNA transposition reaction, but employs orthogonal pairs of split inteins to swap out a designated region of a host protein with an exogenous molecular cassette. We show using isotopic labeling experiments that this 'protein transposition' reaction is concerted when the kinetics for the embedded intein pairs are suitably matched. Critically, this feature allows for efficient manipulation of protein primary structure in the context of a native fold. The utility of this method is illustrated using several protein systems including the multisubunit chromatin remodeling complex, ACF, where we also show protein transposition can occur in situ within the cell nucleus. By carrying out a molecular 'cut and paste' on a protein or protein complex under native folding conditions, our approach dramatically expands the scope of protein semisynthesis.
Conflict of interest statement
Competing interests: The authors declare that they have no competing interests.
Figures



References
-
- Conibear A. C., Watson E. E., Payne R. J., Becker C. F. W., Native chemical ligation in protein synthesis and semi-synthesis. Chem. Soc. Rev. 47, 9046–9068 (2018). - PubMed
-
- Cowieson N. P. et al., An automatable screen for the rapid identification of proteins amenable to refolding. Proteomics 6, 1750–1757 (2006). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources