

Transcriptional immune suppression and up-regulation of double-stranded DNA damage and repair repertoires in ecDNA-containing tumors
- PMID: 38896472
- PMCID: PMC11186631
- DOI: 10.7554/eLife.88895
Transcriptional immune suppression and up-regulation of double-stranded DNA damage and repair repertoires in ecDNA-containing tumors
Abstract
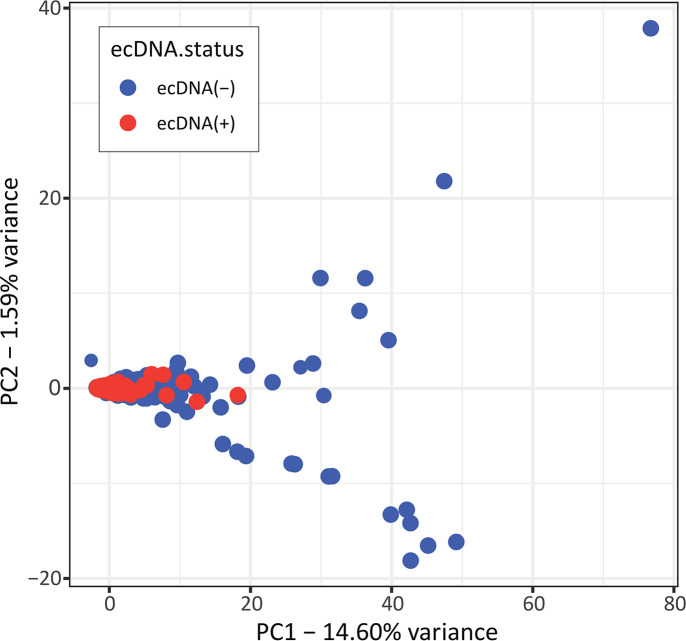
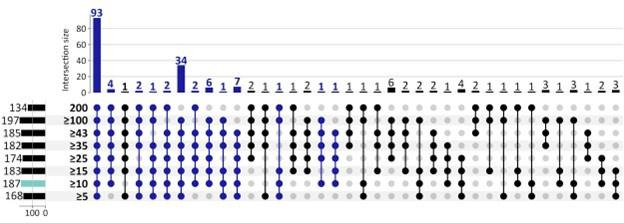
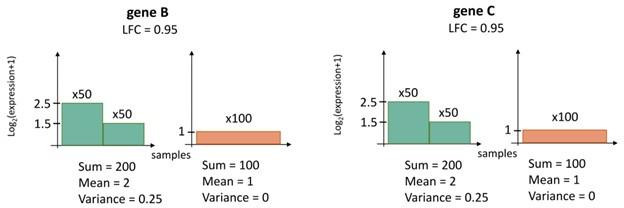
Extrachromosomal DNA is a common cause of oncogene amplification in cancer. The non-chromosomal inheritance of ecDNA enables tumors to rapidly evolve, contributing to treatment resistance and poor outcome for patients. The transcriptional context in which ecDNAs arise and progress, including chromosomally-driven transcription, is incompletely understood. We examined gene expression patterns of 870 tumors of varied histological types, to identify transcriptional correlates of ecDNA. Here, we show that ecDNA-containing tumors impact four major biological processes. Specifically, ecDNA-containing tumors up-regulate DNA damage and repair, cell cycle control, and mitotic processes, but down-regulate global immune regulation pathways. Taken together, these results suggest profound alterations in gene regulation in ecDNA-containing tumors, shedding light on molecular processes that give rise to their development and progression.
Keywords: cancer; chromosomes; ecDNA; extrachromosomal DNA; gene expression; human; transcriptomics.
© 2023, Lin et al.
Conflict of interest statement
ML, SJ No competing interests declared, JL J.L. receives compensation as a consultant for Boundless Bio, HC Reviewing editor, eLife, SW S. Wu is a member of the scientific advisory board of Dimension Genomics Inc, PM P.S.M. is a co-founder and advisor of Boundless Bio. J.L. receives compensation as a consultant for Boundless Bio, VB V.B. is a co-founder, paid consultant, SAB member and has equity interest in Boundless Bio, Inc and Abterra Biosciences, Inc
Figures


























Update of
-
Transcriptional immune suppression and upregulation of double stranded DNA damage and repair repertoires in ecDNA-containing tumors.bioRxiv [Preprint]. 2023 Nov 28:2023.04.24.537925. doi: 10.1101/2023.04.24.537925. bioRxiv. 2023. Update in: Elife. 2024 Jun 19;12:RP88895. doi: 10.7554/eLife.88895. PMID: 37162993 Free PMC article. Updated. Preprint.
References
-
- Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, Osokin N, Kozlov I, Frenkel F, Gancharova O, Almog N, Tsiper M, Ataullakhanov R, Fowler N. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:845–865. doi: 10.1016/j.ccell.2021.04.014. - DOI - PubMed
-
- Bergstrom EN, Luebeck J, Petljak M, Khandekar A, Barnes M, Zhang T, Steele CD, Pillay N, Landi MT, Bafna V, Mischel PS, Harris RS, Alexandrov LB. Mapping clustered mutations in cancer reveals APOBEC3 mutagenesis of ecDNA. Nature. 2022;602:510–517. doi: 10.1038/s41586-022-04398-6. - DOI - PMC - PubMed
MeSH terms
Grants and funding
- OT2CA278635/CA/NCI NIH HHS/United States
- CGCATF-2021/100025/CRUK_/Cancer Research UK/United Kingdom
- U24 CA264379/CA/NCI NIH HHS/United States
- OT2CA278683/CA/NCI NIH HHS/United States
- RR210034/Cancer Prevention and Research Institute of Texas
- OT2 CA278635/CA/NCI NIH HHS/United States
- R01GM114362/NH/NIH HHS/United States
- OT2 CA278688/CA/NCI NIH HHS/United States
- CGCATF-2021/100012/CRUK_/Cancer Research UK/United Kingdom
- R01 GM114362/GM/NIGMS NIH HHS/United States
- CGCATF-2021/100023/CRUK_/Cancer Research UK/United Kingdom
- U24CA264379/CA/NCI NIH HHS/United States
- HI19C1330/Korea Health Industry Development Institute/Republic of Korea
- OT2 CA278683/CA/NCI NIH HHS/United States
- OT2CA278688/CA/NCI NIH HHS/United States
