

Crosstalk of MAP3K1 and EGFR signaling mediates gene-environment interactions that block developmental tissue closure
- PMID: 38897570
- PMCID: PMC11294703
- DOI: 10.1016/j.jbc.2024.107486
Crosstalk of MAP3K1 and EGFR signaling mediates gene-environment interactions that block developmental tissue closure
Abstract
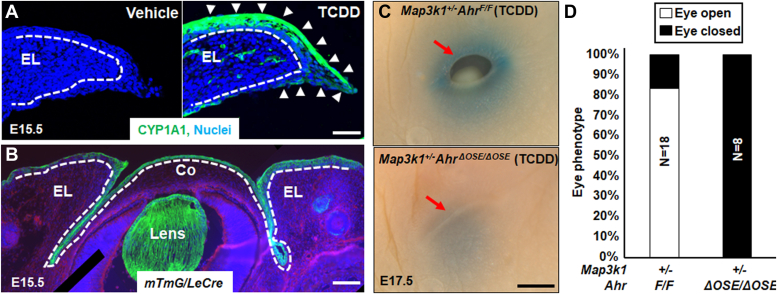
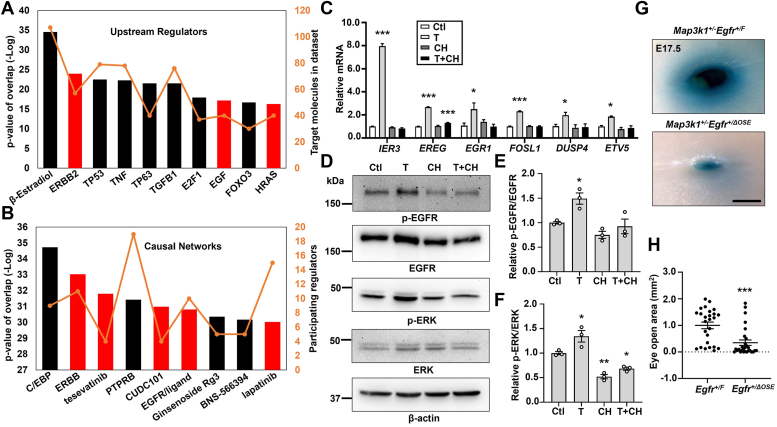
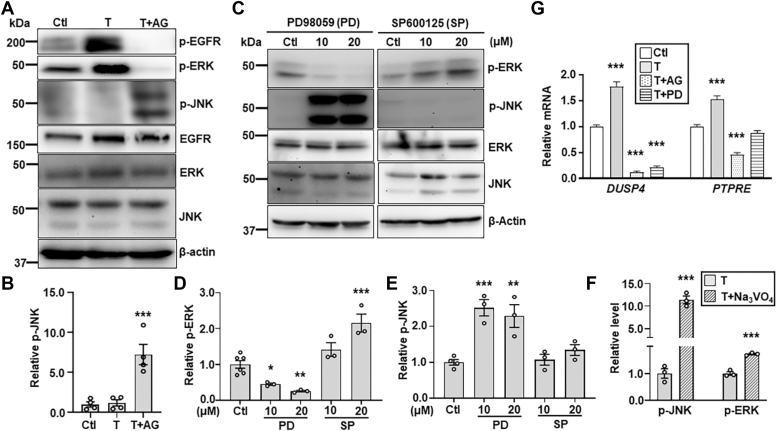
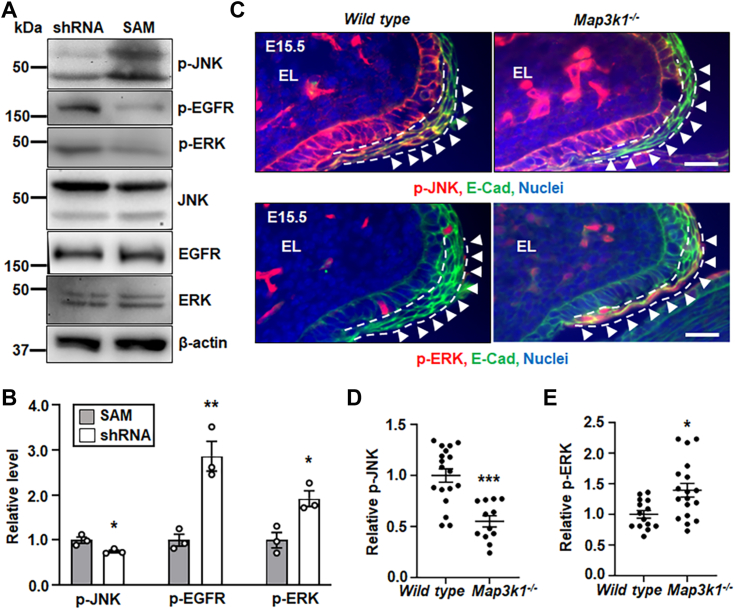
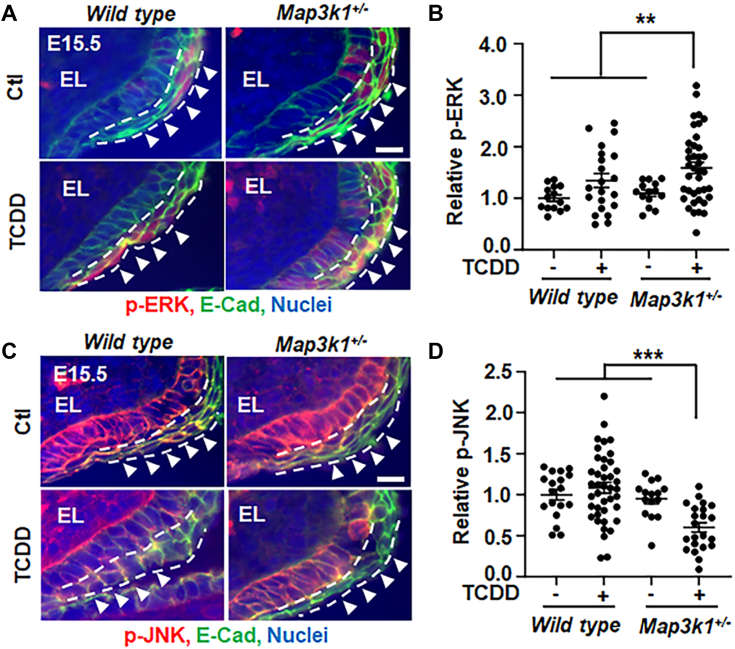
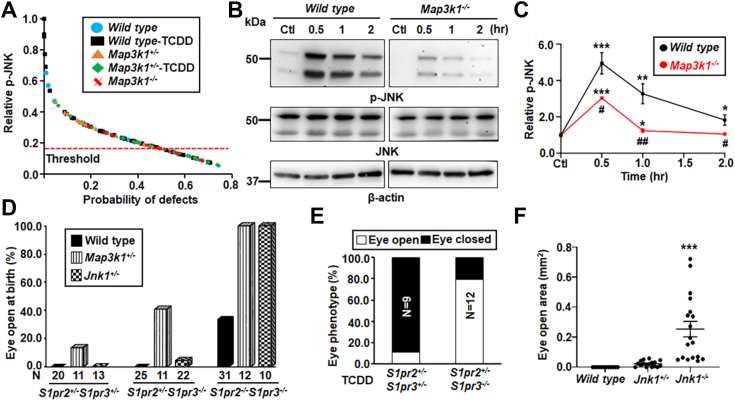
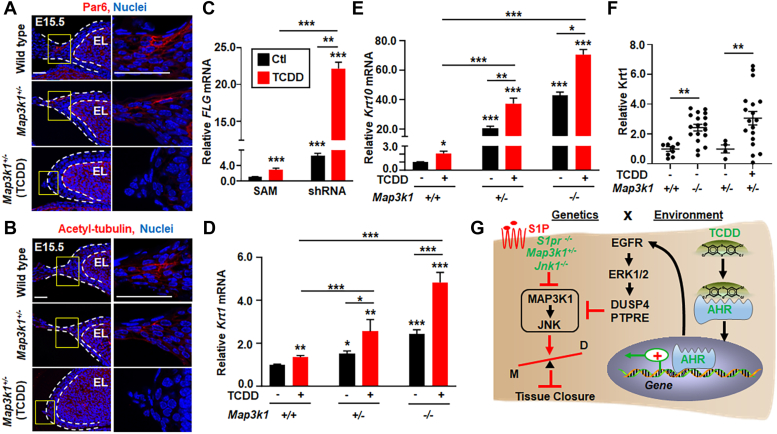
Aberrant regulation of signal transduction pathways can adversely derail biological processes for tissue development. One such process is the embryonic eyelid closure that is dependent on the mitogen-activated protein kinase kinase kinase 1 (MAP3K1). Map3k1 KO in mice results in defective eyelid closure and an autosomal recessive eye-open at birth phenotype. We have shown that in utero exposure to dioxin, a persistent environmental toxicant, induces the same eye defect in Map3k1+/- heterozygous but not WT pups. Here, we explore the mechanisms of the Map3k1 (gene) and dioxin (environment) interactions (GxE) underlying defective eyelid closure. We show that, acting through the aryl hydrocarbon receptor, dioxin activates epidermal growth factor receptor signaling, which in turn depresses MAP3K1-dependent Jun N-terminal kinase (JNK) activity. The dioxin-mediated JNK repression is moderate but is exacerbated by Map3k1 heterozygosity. Therefore, dioxin exposed Map3k1+/- embryonic eyelids have a marked reduction of JNK activity, accelerated differentiation and impeded polarization in the epithelial cells. Knocking out Ahr or Egfr in eyelid epithelium attenuates the open-eye defects in dioxin-treated Map3k1+/- pups, whereas knockout of Jnk1 and S1pr that encodes the sphigosin-1-phosphate (S1P) receptors upstream of the MAP3K1-JNK pathway potentiates the dioxin toxicity. Our novel findings show that the crosstalk of aryl hydrocarbon receptor, epidermal growth factor receptor, and S1P-MAP3K1-JNK pathways determines the outcome of dioxin exposure. Thus, gene mutations targeting these pathways are potential risk factors for the toxicity of environmental chemicals.
Keywords: AHR; EGFR; developmental tissue closure; dioxin; epithelial morphogenesis; gene-environment interactions; the S1PR-MAP3K1-JNK pathway.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Kuida K., Boucher D.M. Functions of MAP kinases: insights from gene-targeting studies. J. Biochem. 2004;135:653–656. - PubMed
-
- Hagemann C., Blank J.L. The ups and downs of MEK kinase interactions. Cell Signal. 2001;13:863–875. - PubMed
-
- Uhlik M.T., Abell A.N., Cuevas B.D., Nakamura K., Johnson G.L. Wiring diagrams of MAPK regulation by MEKK1, 2, and 3. Biochem. Cell Biol. 2004;82:658–663. - PubMed
-
- Craig E.A., Stevens M.V., Vaillancourt R.R., Camenisch T.D. MAP3Ks as central regulators of cell fate during development. Dev. Dyn. 2008;237:3102–3114. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
