

Human SARS-CoV-2 challenge uncovers local and systemic response dynamics
- PMID: 38898278
- PMCID: PMC11222146
- DOI: 10.1038/s41586-024-07575-x
Human SARS-CoV-2 challenge uncovers local and systemic response dynamics
Erratum in
-
Author Correction: Human SARS-CoV-2 challenge uncovers local and systemic response dynamics.Nature. 2024 Aug;632(8025):E3. doi: 10.1038/s41586-024-07838-7. Nature. 2024. PMID: 39090314 Free PMC article. No abstract available.
Abstract
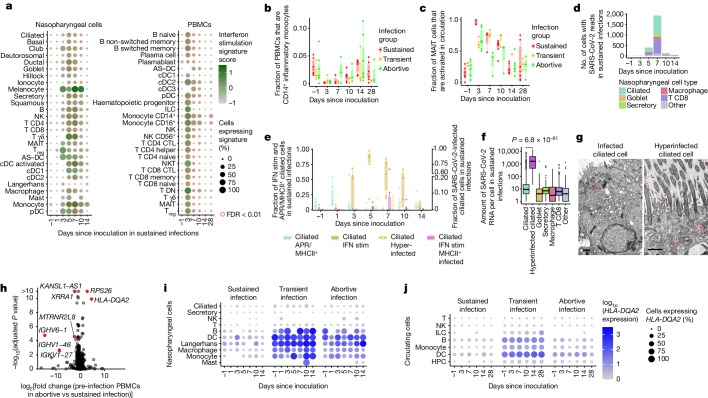
The COVID-19 pandemic is an ongoing global health threat, yet our understanding of the dynamics of early cellular responses to this disease remains limited1. Here in our SARS-CoV-2 human challenge study, we used single-cell multi-omics profiling of nasopharyngeal swabs and blood to temporally resolve abortive, transient and sustained infections in seronegative individuals challenged with pre-Alpha SARS-CoV-2. Our analyses revealed rapid changes in cell-type proportions and dozens of highly dynamic cellular response states in epithelial and immune cells associated with specific time points and infection status. We observed that the interferon response in blood preceded the nasopharyngeal response. Moreover, nasopharyngeal immune infiltration occurred early in samples from individuals with only transient infection and later in samples from individuals with sustained infection. High expression of HLA-DQA2 before inoculation was associated with preventing sustained infection. Ciliated cells showed multiple immune responses and were most permissive for viral replication, whereas nasopharyngeal T cells and macrophages were infected non-productively. We resolved 54 T cell states, including acutely activated T cells that clonally expanded while carrying convergent SARS-CoV-2 motifs. Our new computational pipeline Cell2TCR identifies activated antigen-responding T cells based on a gene expression signature and clusters these into clonotype groups and motifs. Overall, our detailed time series data can serve as a Rosetta stone for epithelial and immune cell responses and reveals early dynamic responses associated with protection against infection.
© 2024. The Author(s).
Conflict of interest statement
R.G.H.L., L.M.D., R.E. and S.A.T. are inventors on a filed patent that is related to the detection and application of activated T cells. In the past 3 years, S.A.T. has received remuneration for scientific advisory board membership from Sanofi, GlaxoSmithKline, Foresite Labs and Qiagen. S.A.T. is a co-founder and holds equity in Transition Bio and Ensocell. From 8 January 2024, S.A.T. is a part-time employee of GlaxoSmithKline. R.E. is a co-founder and equity holder in Ensocell. P.M. is a Medical Research Council (MRC)-GlaxoSmithKline EMINENT clinical training fellow with project funding unrelated to the topic of this work and receives co-funding from the National Institute for Health Research (NIHR) University College London Hospitals (UCLH) Biomedical Research Centre. P.M. reports consultancy fees from SOBI, AbbVie, UCB, Lilly, Boehringer Ingelheim and EUSA Pharma, all unrelated to this study. S.M.J. has received fees for advisory board membership in the last three years from Bard1 Lifescience. He has received grant income from GRAIL Inc. He is an unpaid member of a GRAIL advisory board. He has received lecture fees for academic meetings from Cheisi and AstraZeneca. His wife works for AstraZeneca. R.C.C. has research collaborations with Chiesi Chiesi Farmaceutici S.p.A. and GSK and receives consulting fees from Vicore, outside the submitted work. A. Mann, A.C., M.K., M.M. and A.B. are full-time employees at hVIVO Services. The other authors declare no competing interests.
Figures













References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
