

Computational design of soluble and functional membrane protein analogues
- PMID: 38898281
- PMCID: PMC11236705
- DOI: 10.1038/s41586-024-07601-y
Computational design of soluble and functional membrane protein analogues
Abstract
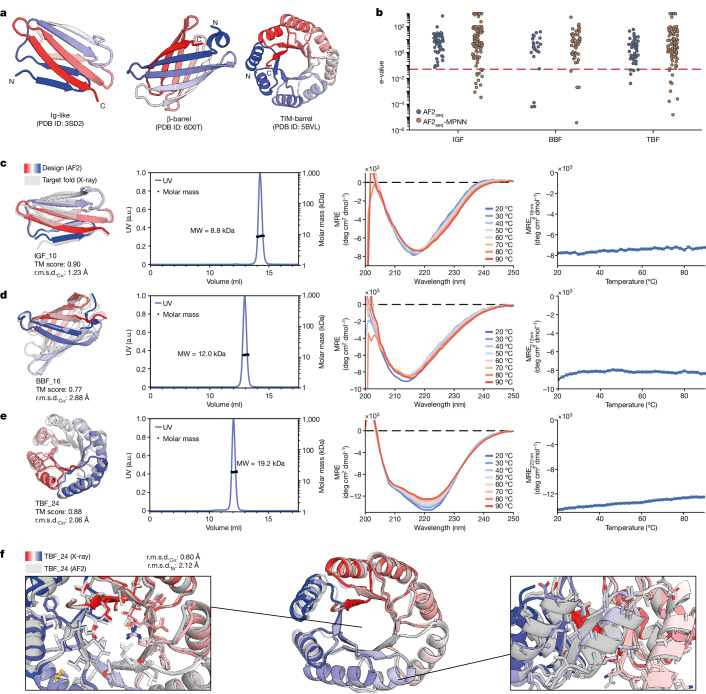
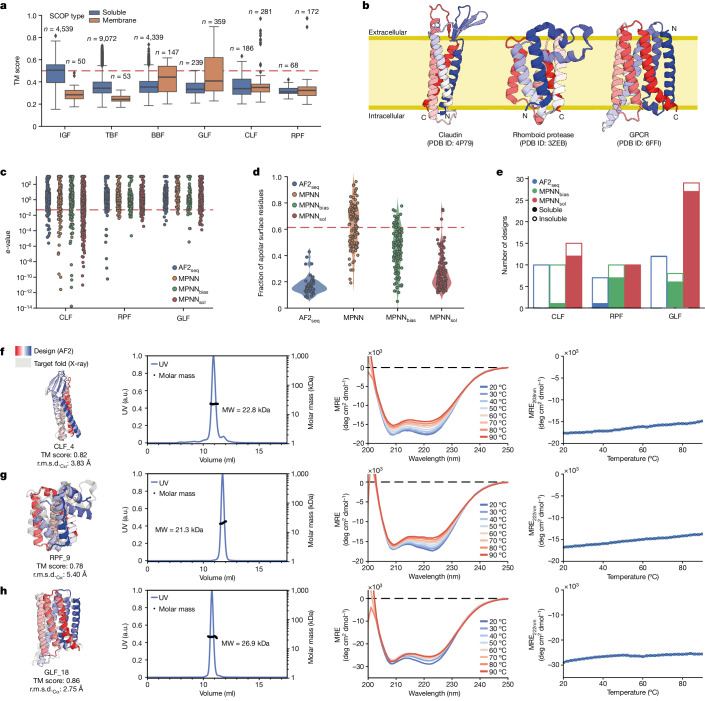
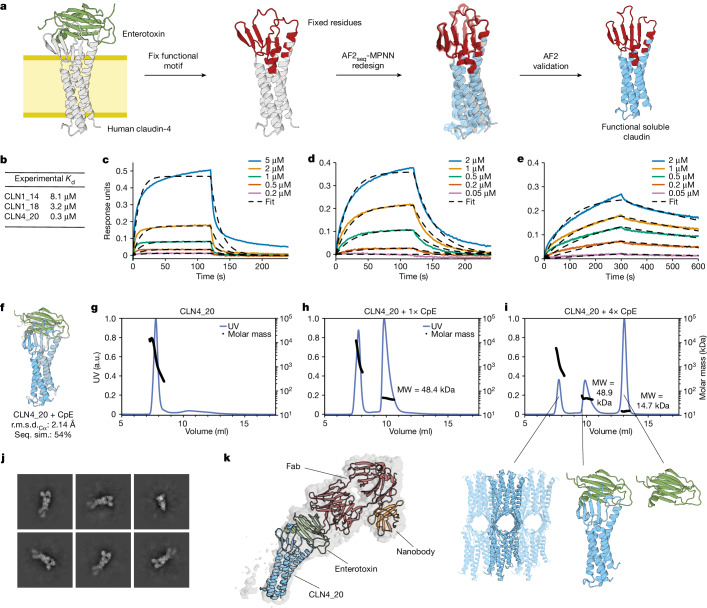
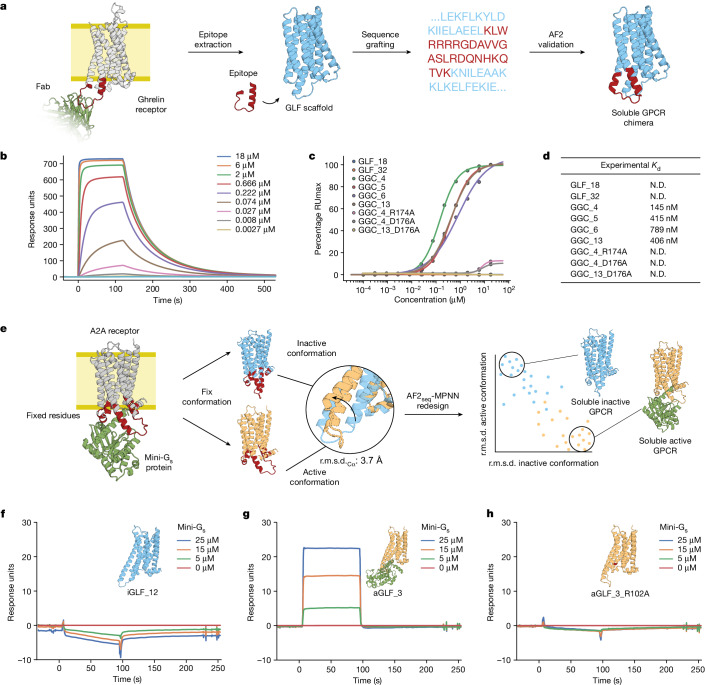
De novo design of complex protein folds using solely computational means remains a substantial challenge1. Here we use a robust deep learning pipeline to design complex folds and soluble analogues of integral membrane proteins. Unique membrane topologies, such as those from G-protein-coupled receptors2, are not found in the soluble proteome, and we demonstrate that their structural features can be recapitulated in solution. Biophysical analyses demonstrate the high thermal stability of the designs, and experimental structures show remarkable design accuracy. The soluble analogues were functionalized with native structural motifs, as a proof of concept for bringing membrane protein functions to the soluble proteome, potentially enabling new approaches in drug discovery. In summary, we have designed complex protein topologies and enriched them with functionalities from membrane proteins, with high experimental success rates, leading to a de facto expansion of the functional soluble fold space.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












Update of
-
Computational design of soluble functional analogues of integral membrane proteins.bioRxiv [Preprint]. 2024 Mar 7:2023.05.09.540044. doi: 10.1101/2023.05.09.540044. bioRxiv. 2024. Update in: Nature. 2024 Jul;631(8020):449-458. doi: 10.1038/s41586-024-07601-y. PMID: 38496615 Free PMC article. Updated. Preprint.
References
-
- Huang, B. et al. A backbone-centred energy function of neural networks for protein design. Nature602, 523–528 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
