

An integrated single-cell RNA-seq map of human neuroblastoma tumors and preclinical models uncovers divergent mesenchymal-like gene expression programs
- PMID: 38898465
- PMCID: PMC11186099
- DOI: 10.1186/s13059-024-03309-4
An integrated single-cell RNA-seq map of human neuroblastoma tumors and preclinical models uncovers divergent mesenchymal-like gene expression programs
Abstract
Background: Neuroblastoma is a common pediatric cancer, where preclinical studies suggest that a mesenchymal-like gene expression program contributes to chemotherapy resistance. However, clinical outcomes remain poor, implying we need a better understanding of the relationship between patient tumor heterogeneity and preclinical models.
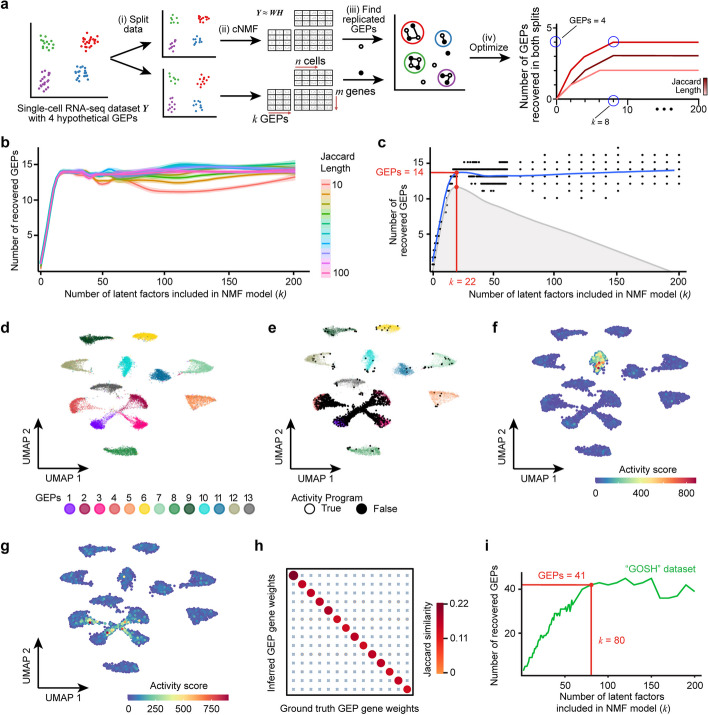
Results: Here, we generate single-cell RNA-seq maps of neuroblastoma cell lines, patient-derived xenograft models (PDX), and a genetically engineered mouse model (GEMM). We develop an unsupervised machine learning approach ("automatic consensus nonnegative matrix factorization" (acNMF)) to compare the gene expression programs found in preclinical models to a large cohort of patient tumors. We confirm a weakly expressed, mesenchymal-like program in otherwise adrenergic cancer cells in some pre-treated high-risk patient tumors, but this appears distinct from the presumptive drug-resistance mesenchymal programs evident in cell lines. Surprisingly, however, this weak-mesenchymal-like program is maintained in PDX and could be chemotherapy-induced in our GEMM after only 24 h, suggesting an uncharacterized therapy-escape mechanism.
Conclusions: Collectively, our findings improve the understanding of how neuroblastoma patient tumor heterogeneity is reflected in preclinical models, provides a comprehensive integrated resource, and a generalizable set of computational methodologies for the joint analysis of clinical and pre-clinical single-cell RNA-seq datasets.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





Update of
-
An integrated single-cell RNA-seq map of human neuroblastoma tumors and preclinical models uncovers divergent mesenchymal-like gene expression programs.bioRxiv [Preprint]. 2024 Apr 22:2023.04.13.536639. doi: 10.1101/2023.04.13.536639. bioRxiv. 2024. Update in: Genome Biol. 2024 Jun 19;25(1):161. doi: 10.1186/s13059-024-03309-4. PMID: 38712039 Free PMC article. Updated. Preprint.
References
-
- Moreno L, et al. Accelerating drug development for neuroblastoma: summary of the second neuroblastoma drug development strategy forum from innovative therapies for children with cancer and International Society of Paediatric Oncology Europe Neuroblastoma. Eur J Cancer. 2020;136:52–68. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
