

Simple and Accurate One-Body Energy and Dipole Moment Surfaces for Water and Beyond
- PMID: 38900596
- PMCID: PMC11229074
- DOI: 10.1021/acs.jpclett.4c00587
Simple and Accurate One-Body Energy and Dipole Moment Surfaces for Water and Beyond
Abstract
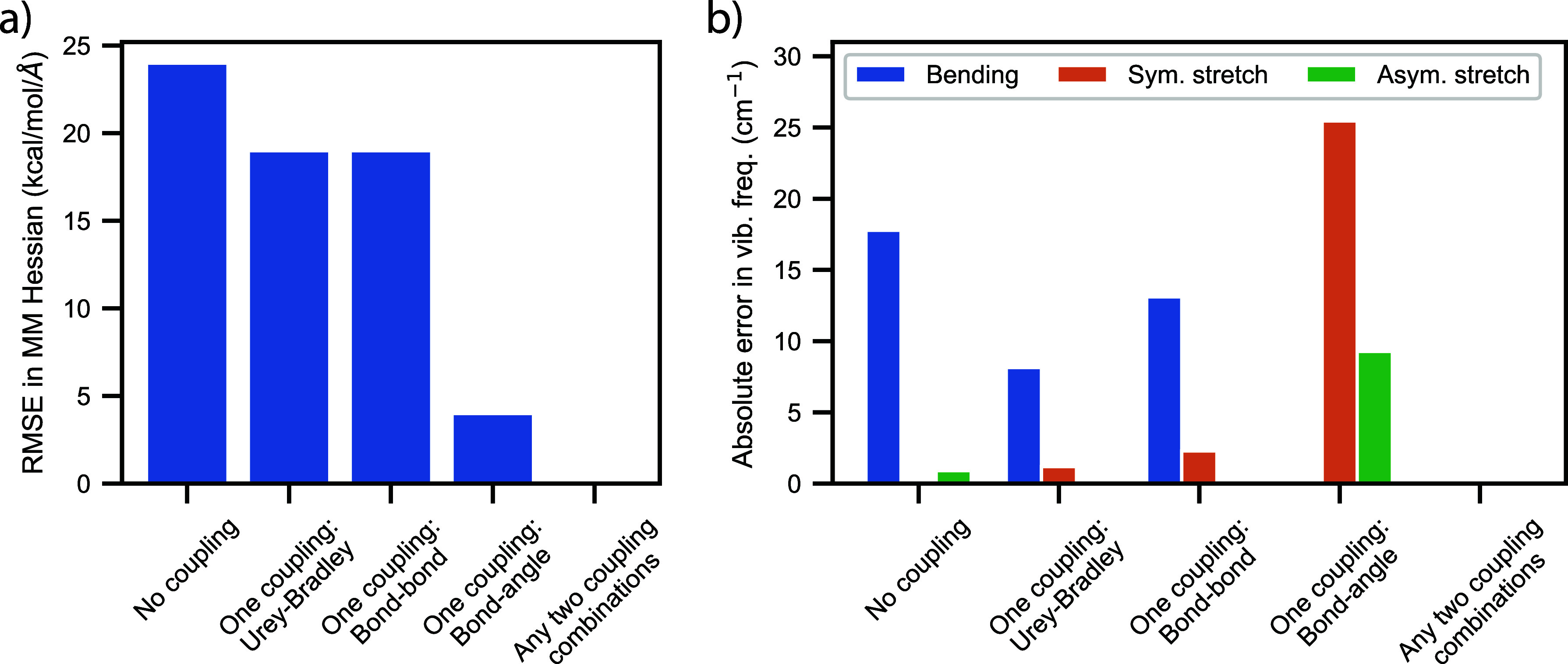
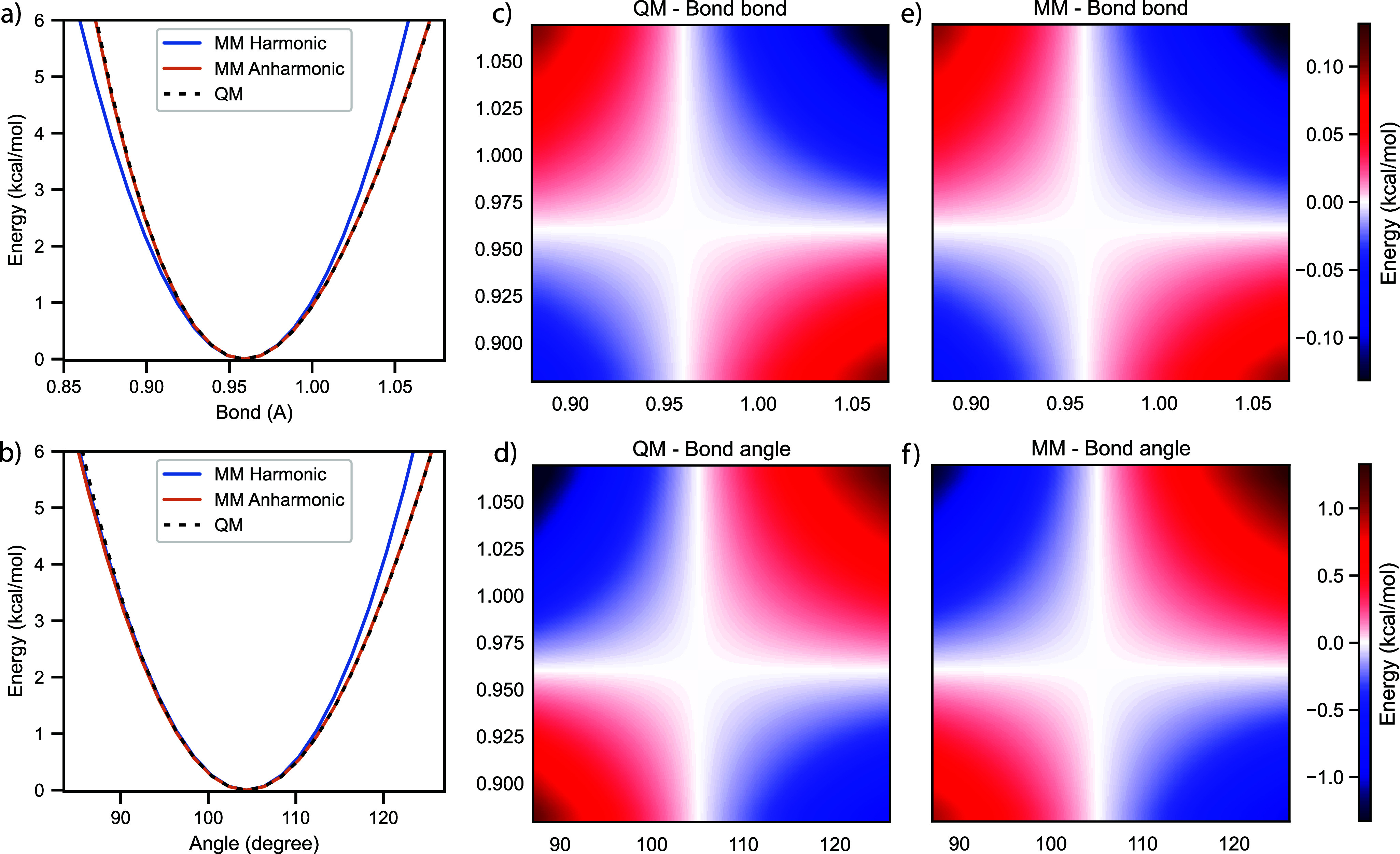
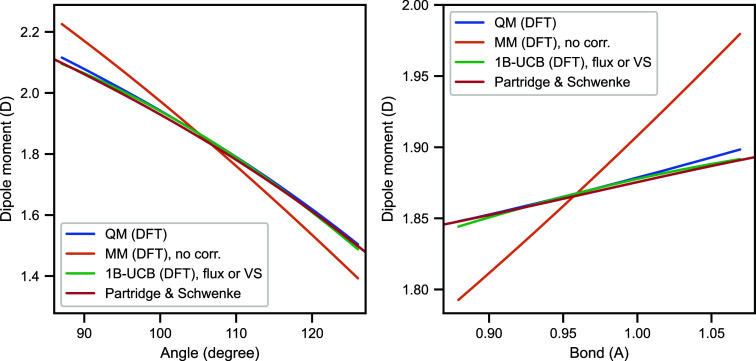
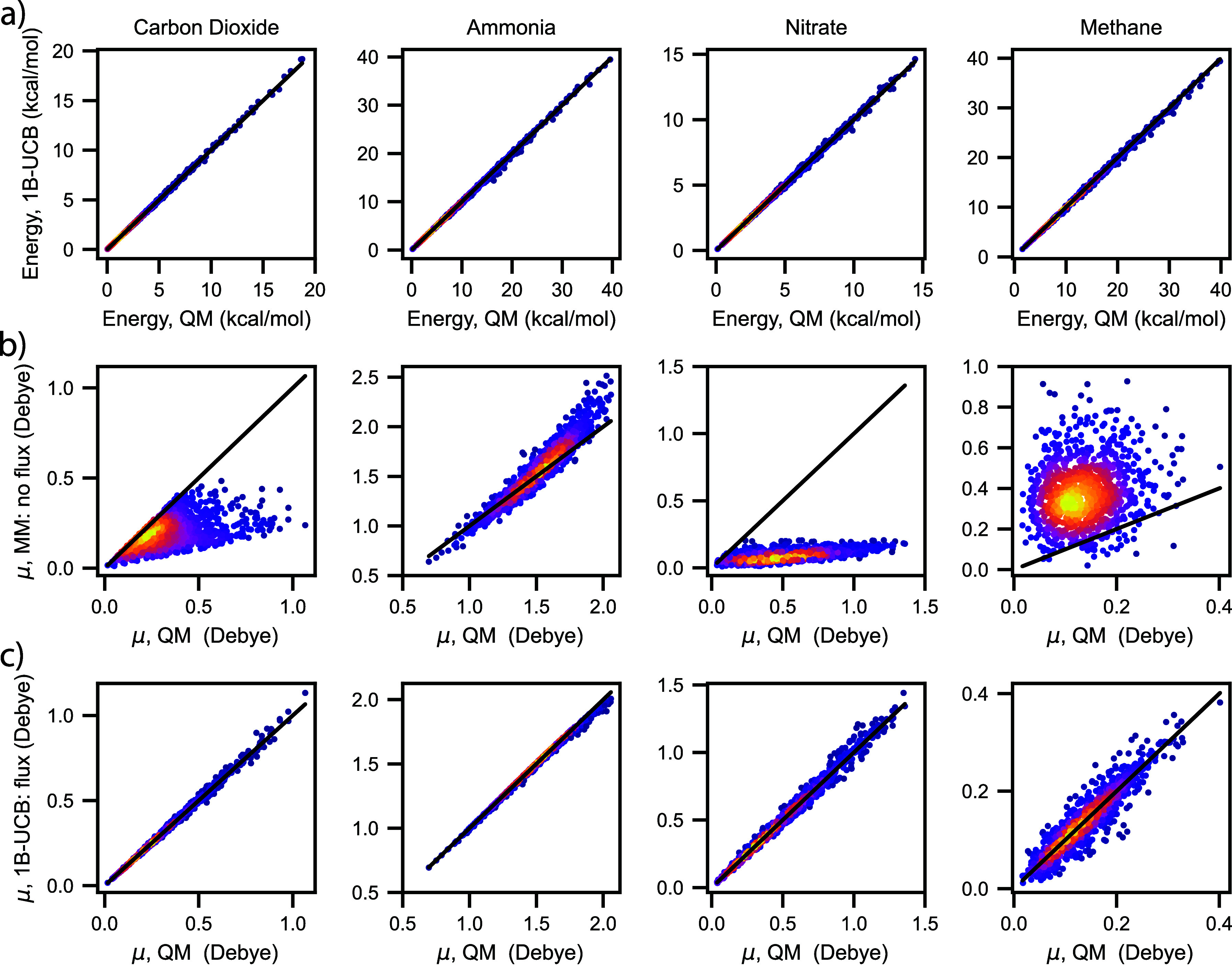
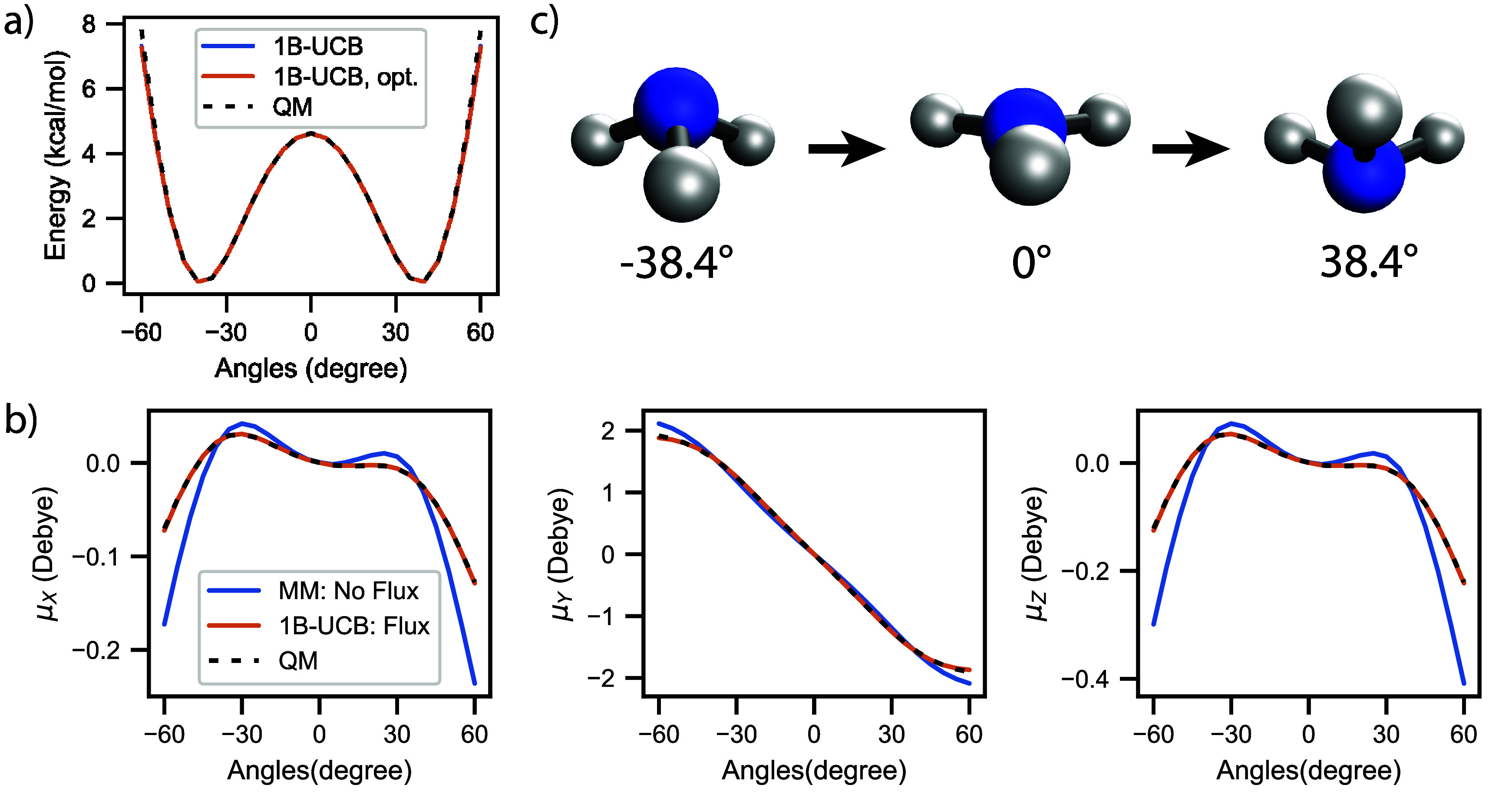
Water is often the testing ground for new, advanced force fields. While advanced functional forms for intermolecular interactions have been integral to the development of accurate water models, less attention has been paid to a transferable model for intramolecular valence terms. In this work, we present a one-body energy and dipole moment surface model, named 1B-UCB, that is simple yet accurate and can be feasibly adapted for both standard and advanced potentials. 1B-UCB for water is comparable in accuracy to those with much more complex functional forms, despite having drastically fewer parameters. The parametrization protocol has been implemented as part of the Q-Force automated workflow and requires only a quantum mechanical Hessian calculation as reference data, hence allowing it to be easily extended to a variety of molecular systems beyond water, which we demonstrate on a selection of small molecules with different symmetries.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

References
-
- Demerdash O.; Wang L.-P.; Head-Gordon T. Advanced models for water simulations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1355. 10.1002/wcms.1355. - DOI
