

G Protein-Coupled Receptors: A Century of Research and Discovery
- PMID: 38900852
- PMCID: PMC11192237
- DOI: 10.1161/CIRCRESAHA.124.323067
G Protein-Coupled Receptors: A Century of Research and Discovery
Abstract
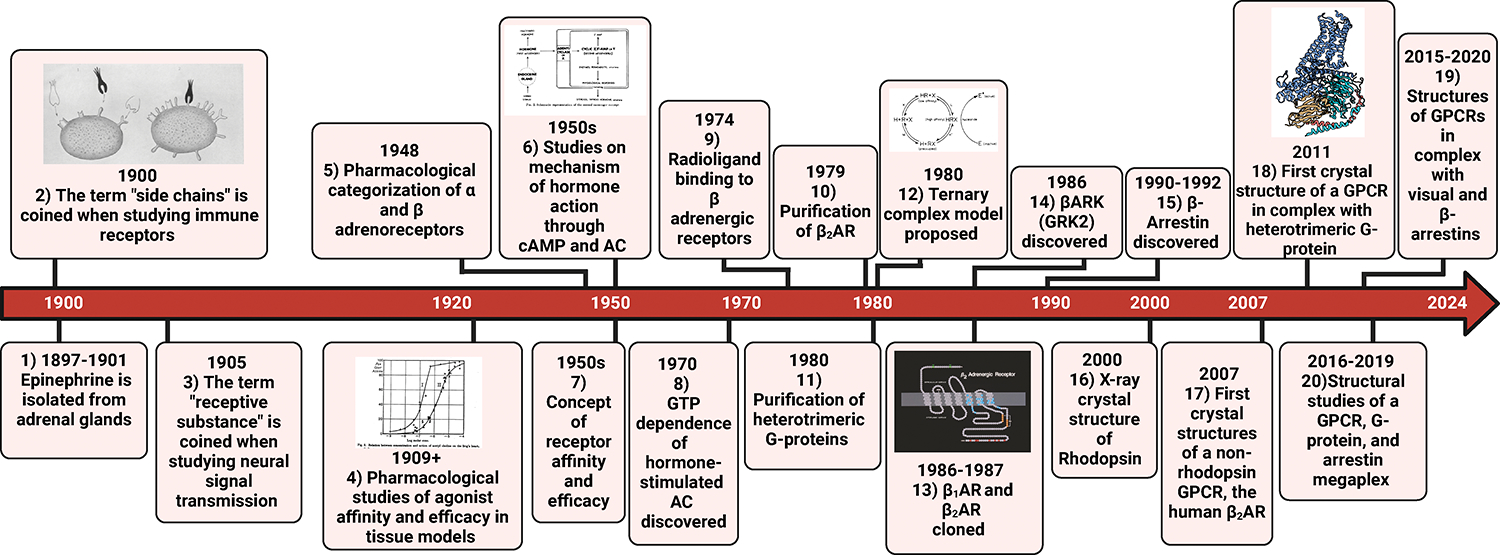
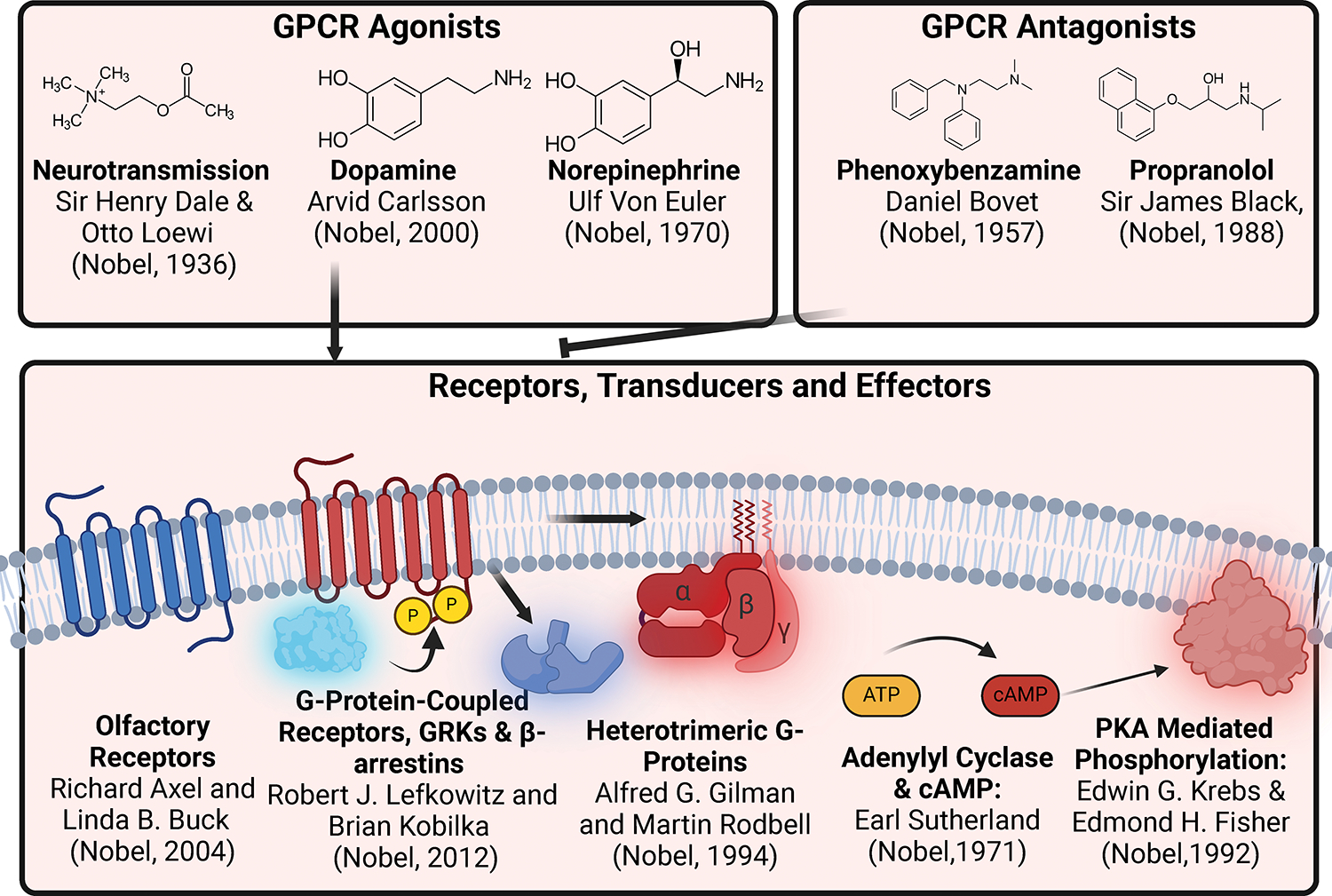
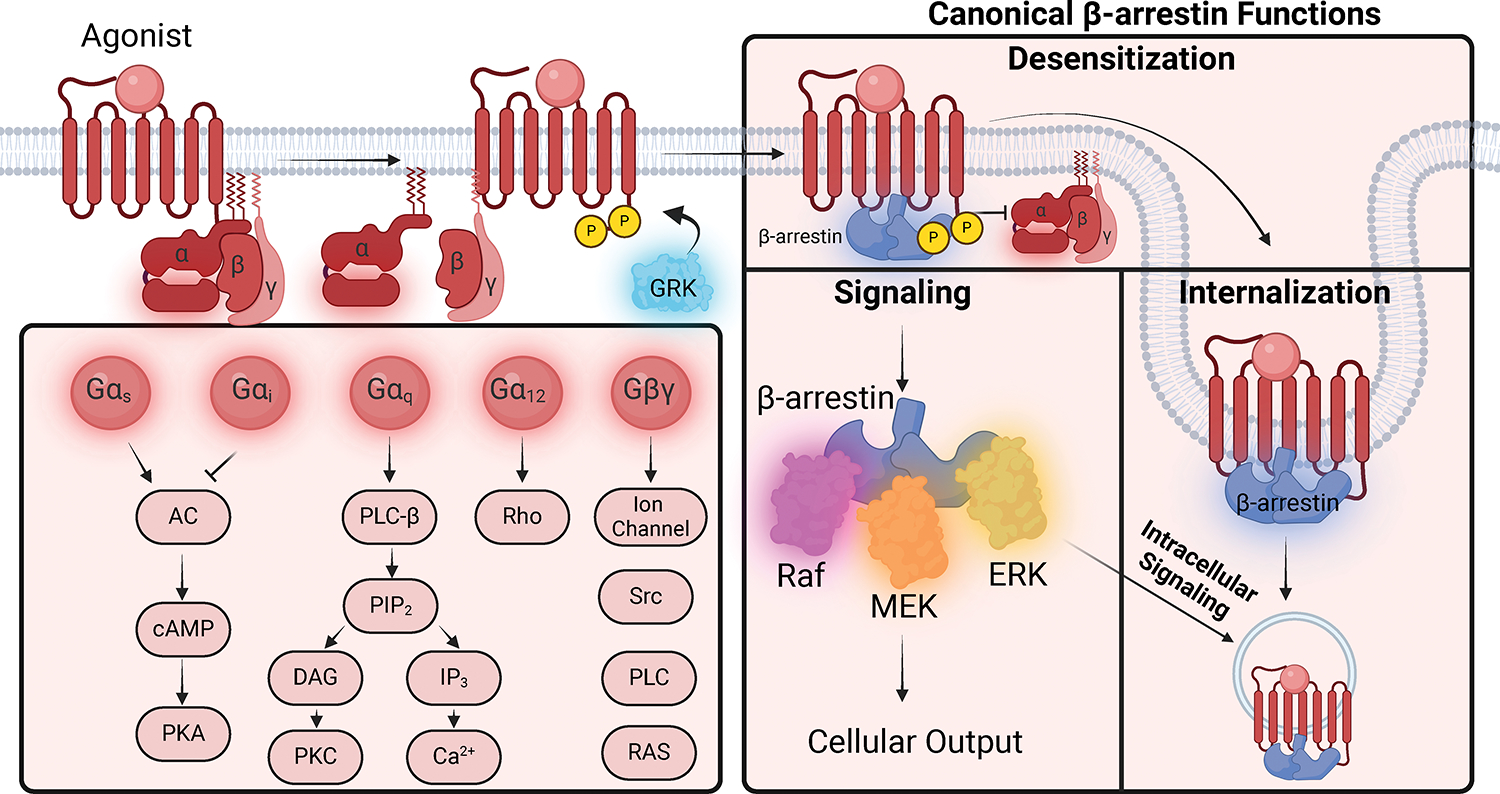
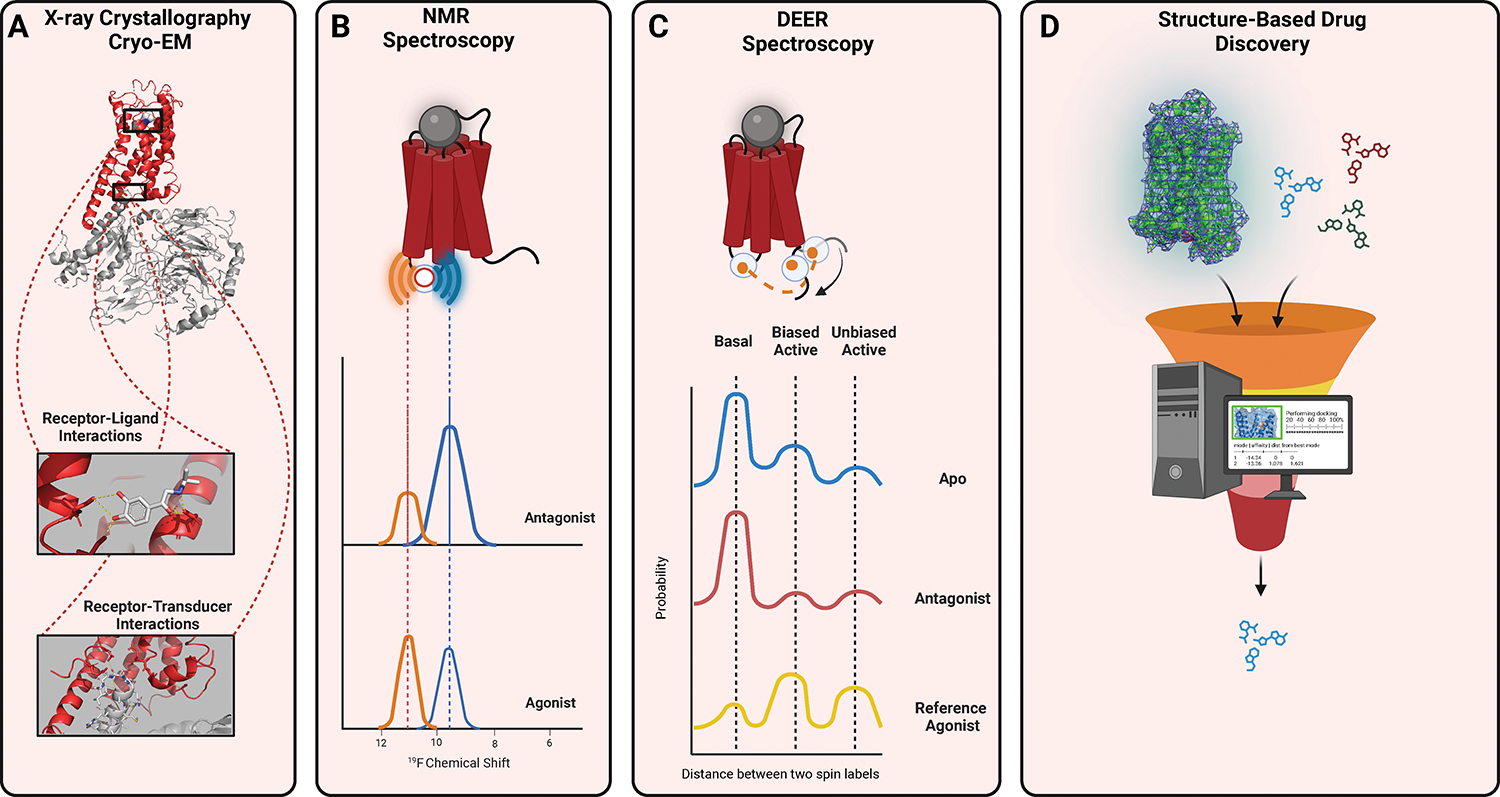
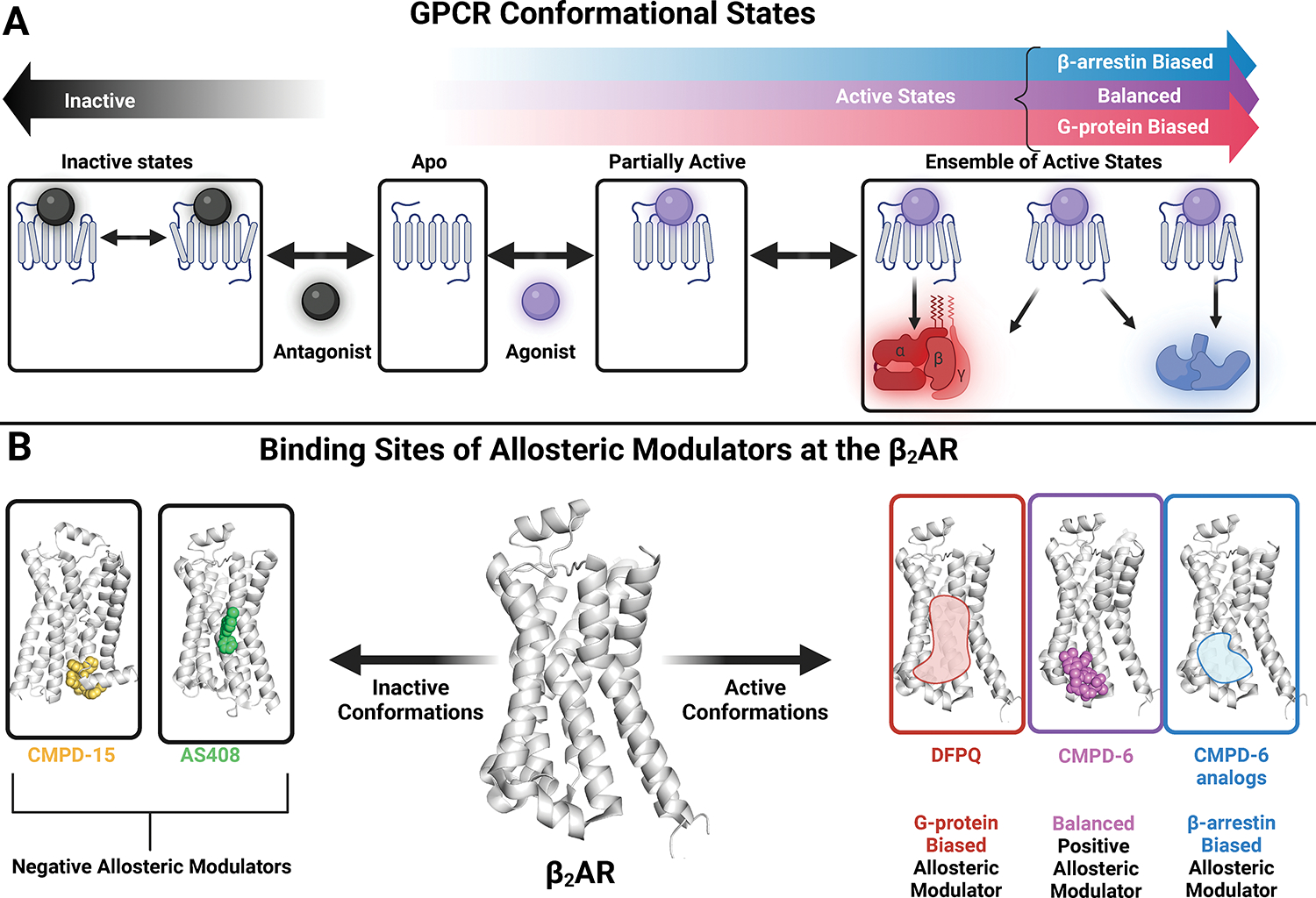
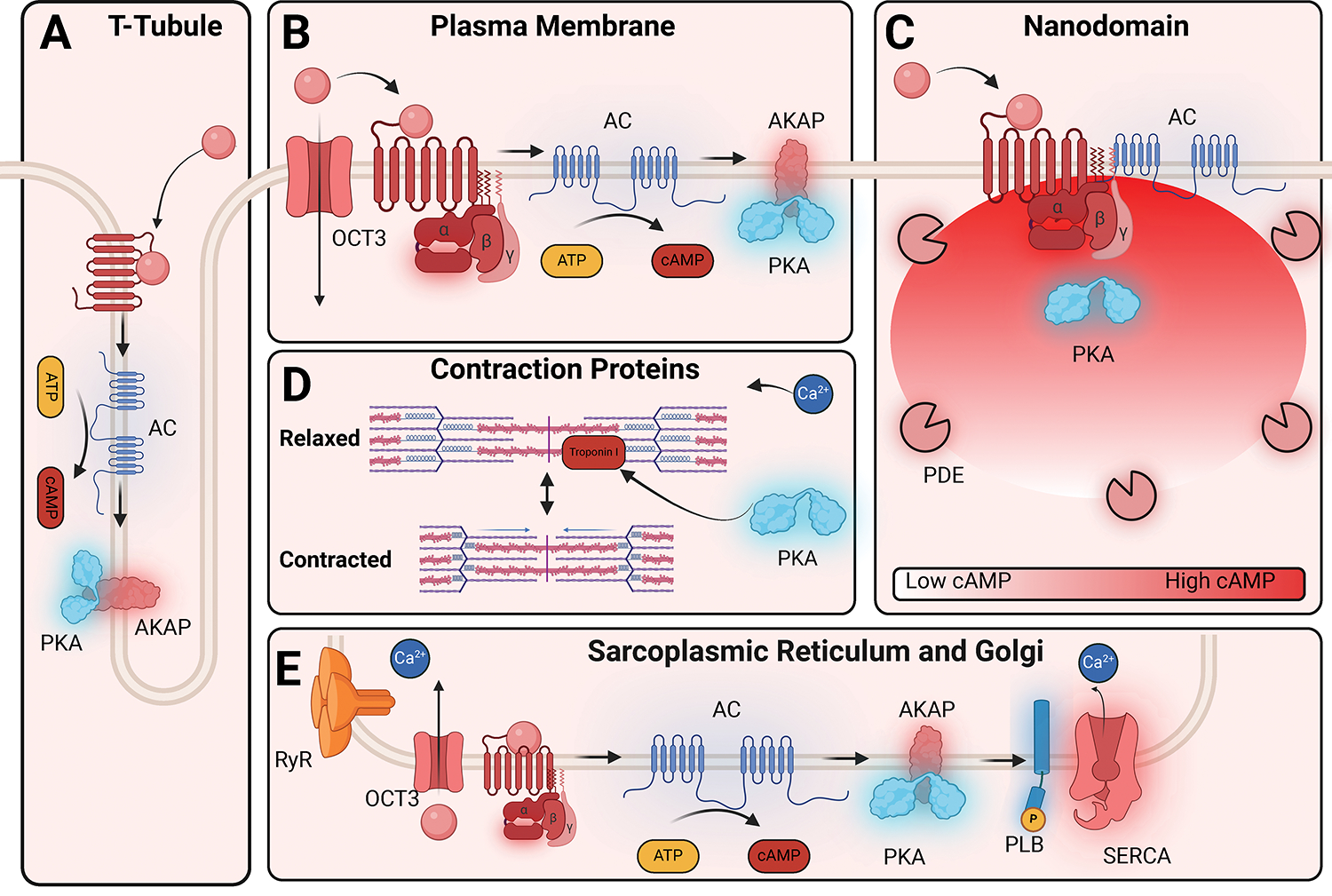
GPCRs (G protein-coupled receptors), also known as 7 transmembrane domain receptors, are the largest receptor family in the human genome, with ≈800 members. GPCRs regulate nearly every aspect of human physiology and disease, thus serving as important drug targets in cardiovascular disease. Sharing a conserved structure comprised of 7 transmembrane α-helices, GPCRs couple to heterotrimeric G-proteins, GPCR kinases, and β-arrestins, promoting downstream signaling through second messengers and other intracellular signaling pathways. GPCR drug development has led to important cardiovascular therapies, such as antagonists of β-adrenergic and angiotensin II receptors for heart failure and hypertension, and agonists of the glucagon-like peptide-1 receptor for reducing adverse cardiovascular events and other emerging indications. There continues to be a major interest in GPCR drug development in cardiovascular and cardiometabolic disease, driven by advances in GPCR mechanistic studies and structure-based drug design. This review recounts the rich history of GPCR research, including the current state of clinically used GPCR drugs, and highlights newly discovered aspects of GPCR biology and promising directions for future investigation. As additional mechanisms for regulating GPCR signaling are uncovered, new strategies for targeting these ubiquitous receptors hold tremendous promise for the field of cardiovascular medicine.
Keywords: adrenergic beta-antagonists; allosteric regulation; angiotensin II; beta-arrestins; g-protein-coupled receptor kinase 2; heart failure; receptors, g-protein-coupled.
Conflict of interest statement
Figures

References
-
- Takamine J Adrenalin the active principle of the suprarenal glands and its mode of preparation. American Journal of Pharmacy (1835–1907). 1901:523.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
