

Structure and repair of replication-coupled DNA breaks
- PMID: 38900911
- PMCID: PMC11620331
- DOI: 10.1126/science.ado3867
Structure and repair of replication-coupled DNA breaks
Abstract
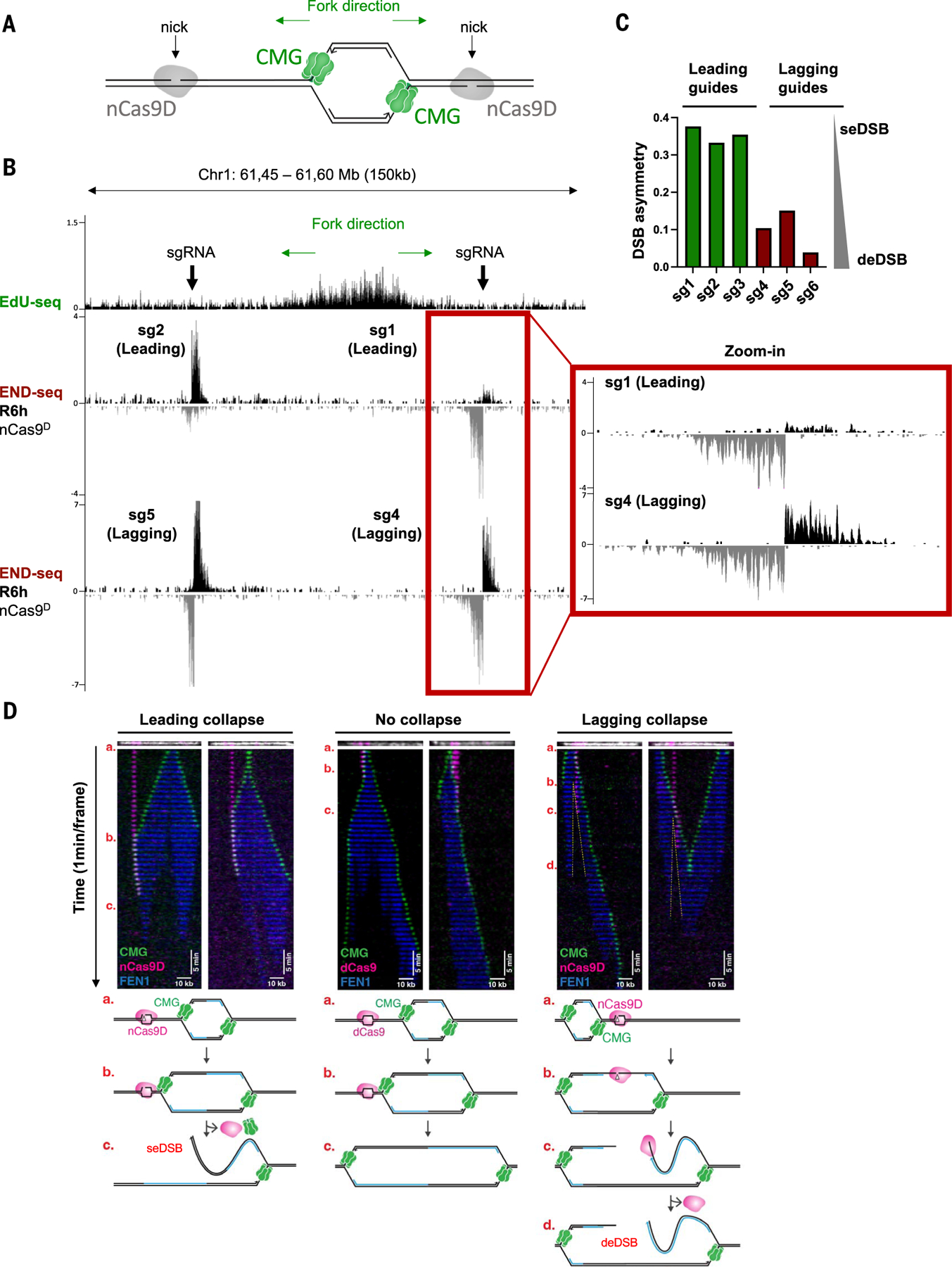
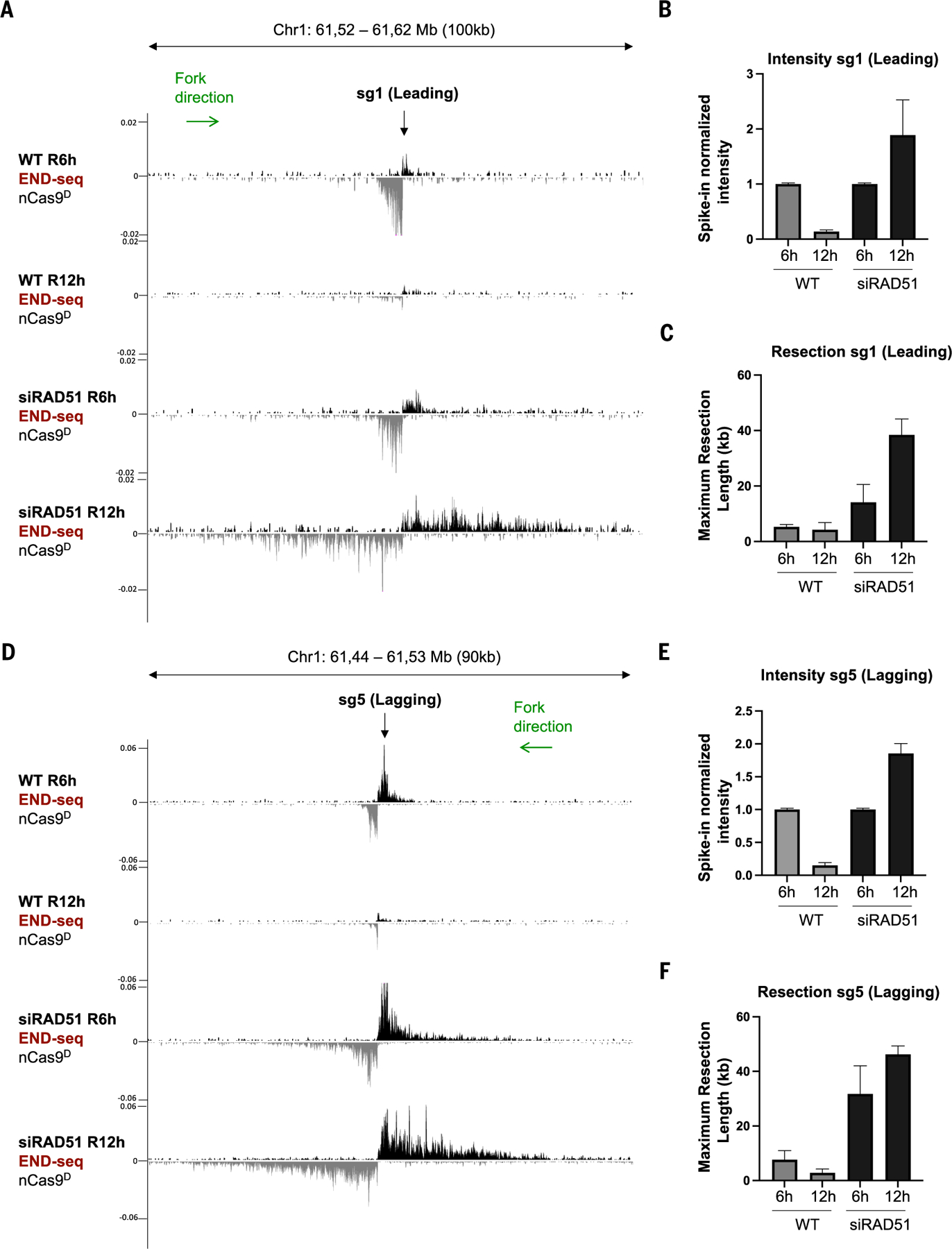
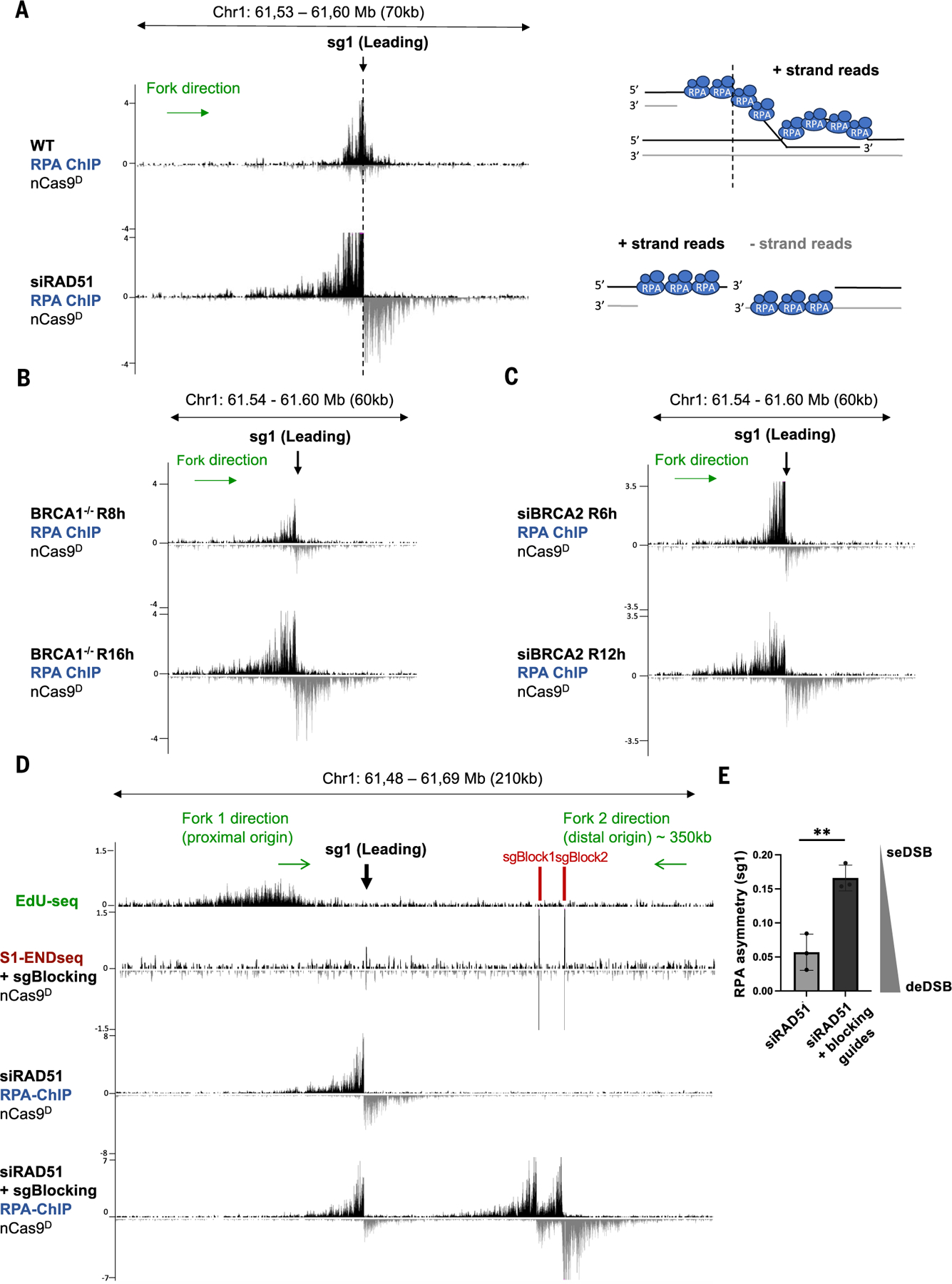
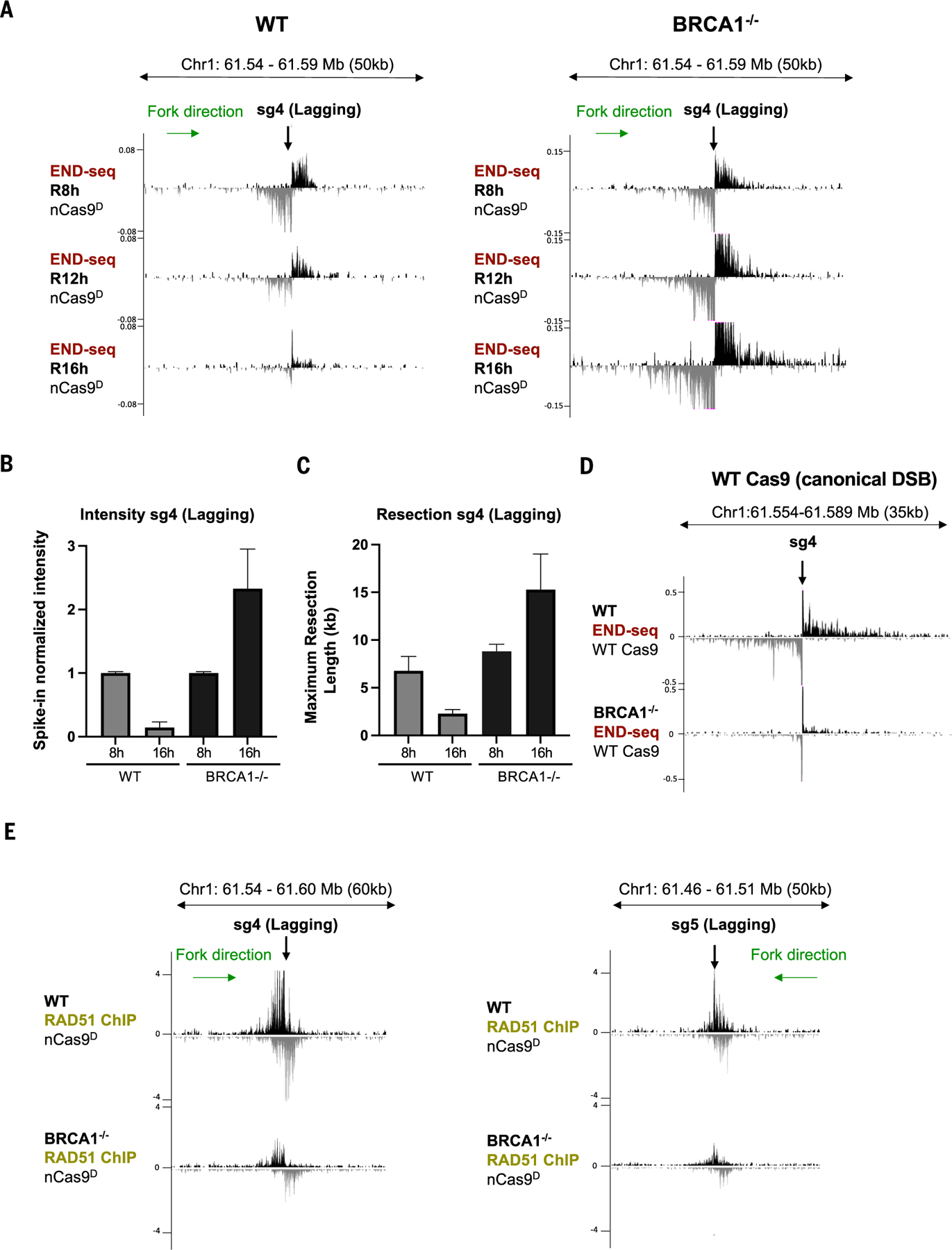
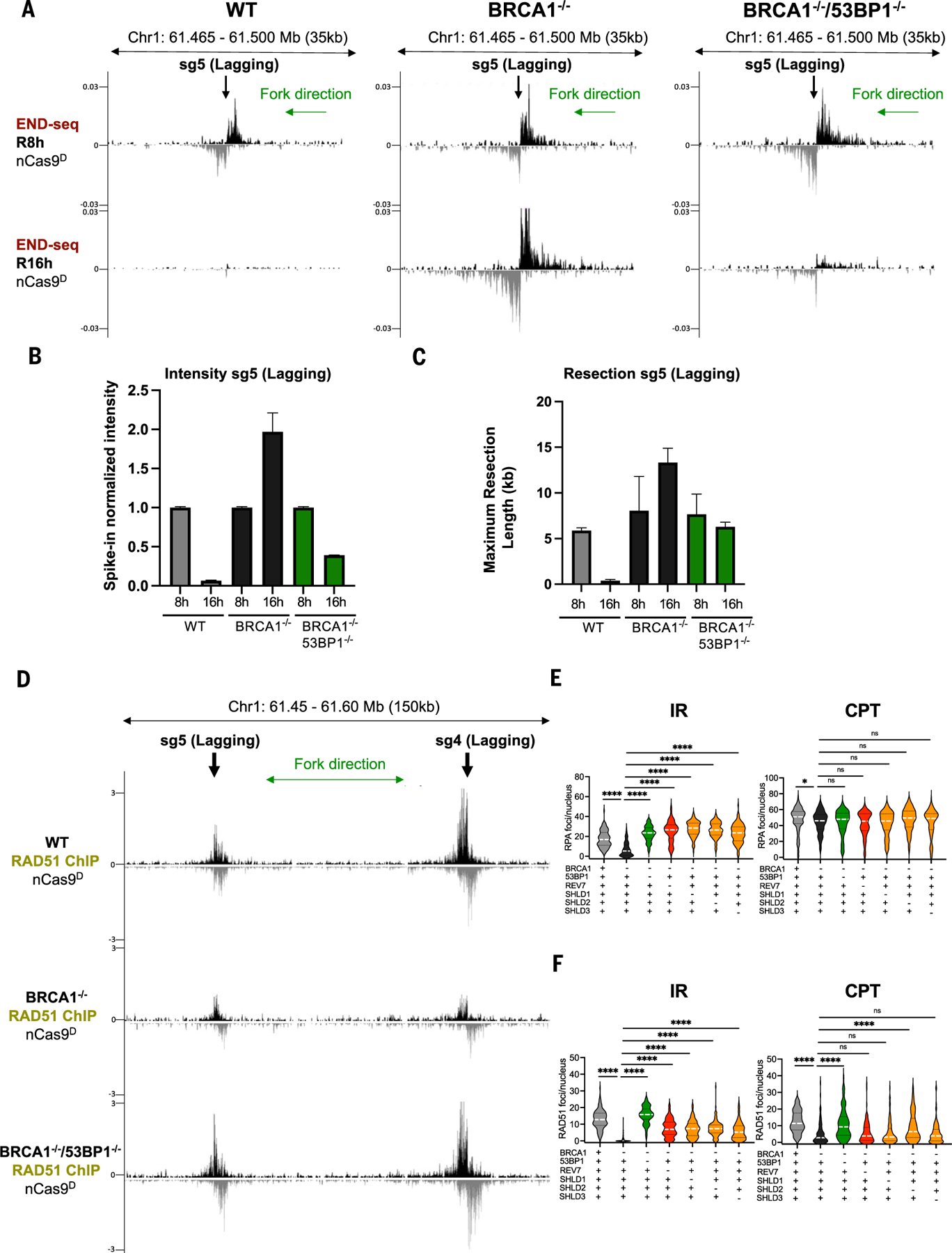
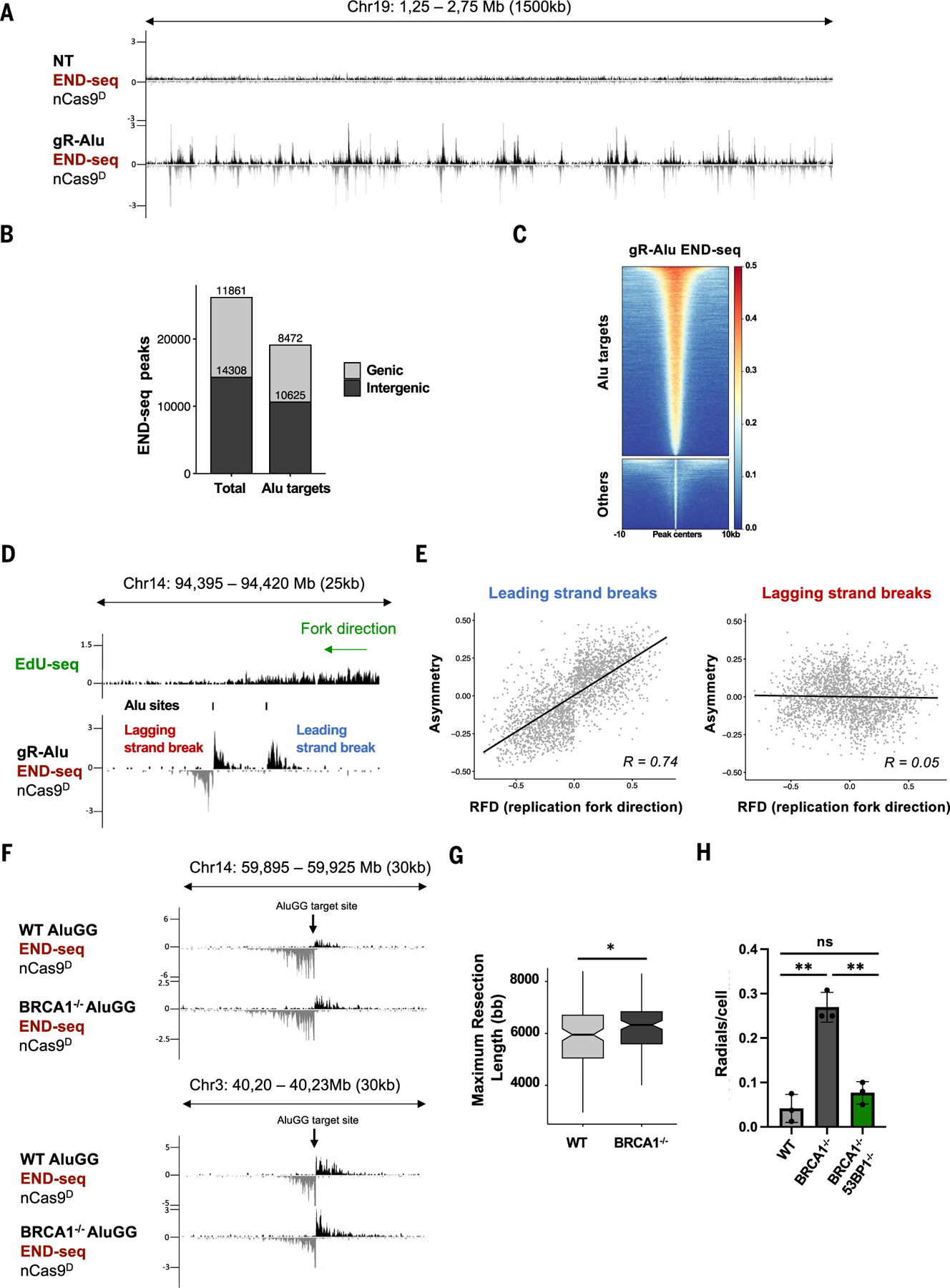
Using CRISPR-Cas9 nicking enzymes, we examined the interaction between the replication machinery and single-strand breaks, one of the most common forms of endogenous DNA damage. We show that replication fork collapse at leading-strand nicks generates resected single-ended double-strand breaks (seDSBs) that are repaired by homologous recombination (HR). If these seDSBs are not promptly repaired, arrival of adjacent forks creates double-ended DSBs (deDSBs), which could drive genomic scarring in HR-deficient cancers. deDSBs can also be generated directly when the replication fork bypasses lagging-strand nicks. Unlike deDSBs produced independently of replication, end resection at nick-induced seDSBs and deDSBs is BRCA1-independent. Nevertheless, BRCA1 antagonizes 53BP1 suppression of RAD51 filament formation. These results highlight distinctive mechanisms that maintain replication fork stability.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
