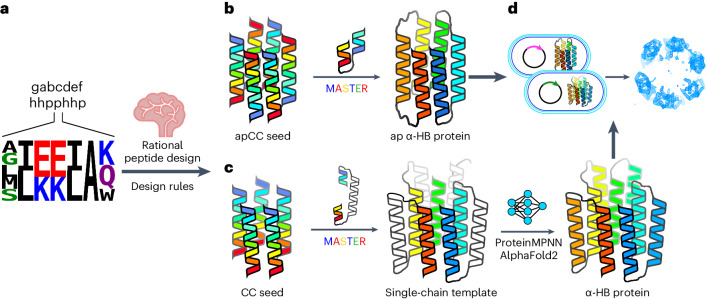
Rationally seeded computational protein design of ɑ-helical barrels
- PMID: 38902458
- PMCID: PMC11288890
- DOI: 10.1038/s41589-024-01642-0
Rationally seeded computational protein design of ɑ-helical barrels
Abstract
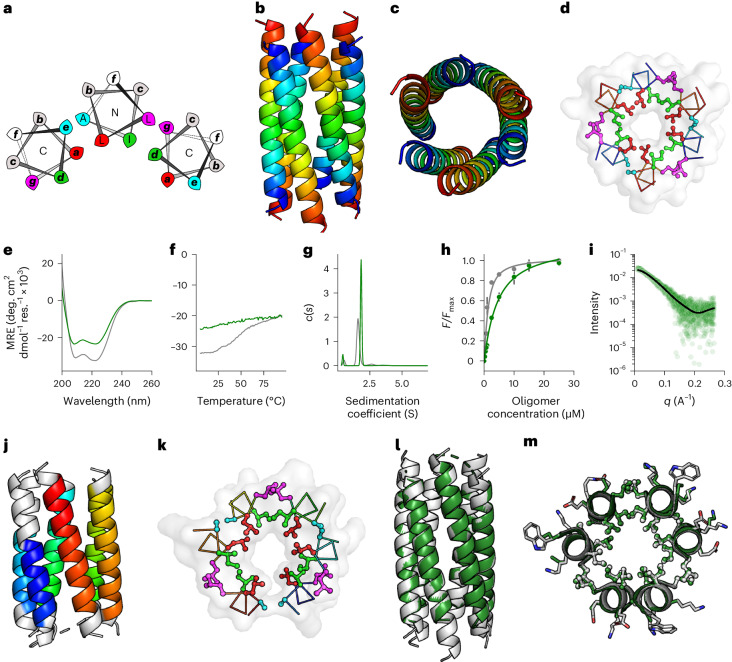
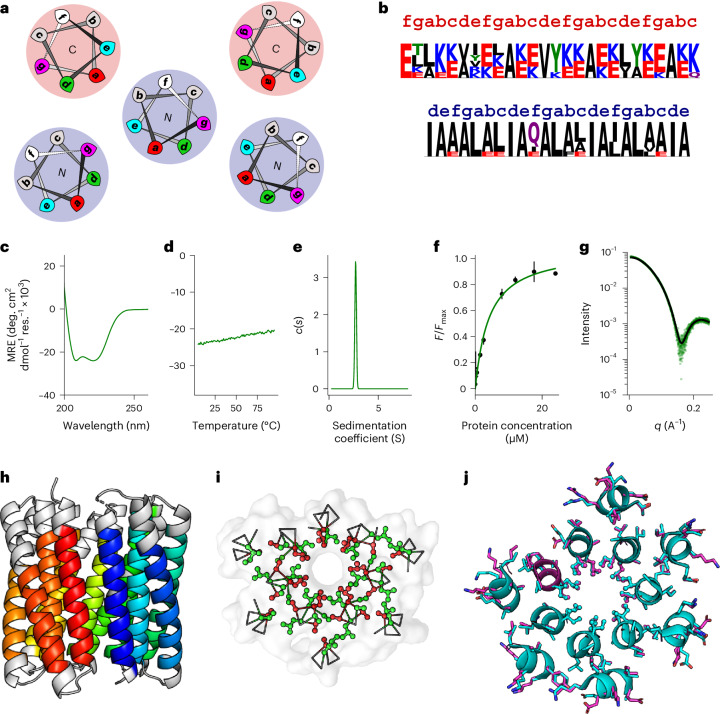
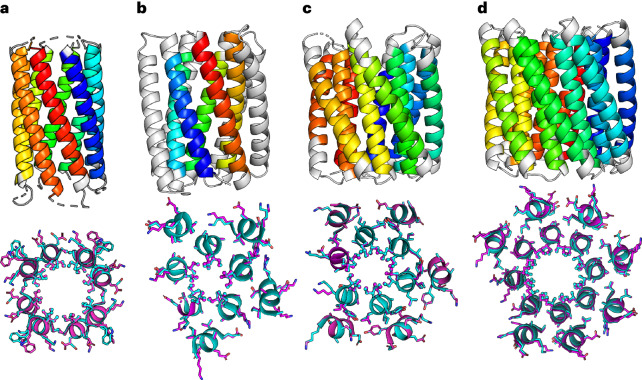
Computational protein design is advancing rapidly. Here we describe efficient routes starting from validated parallel and antiparallel peptide assemblies to design two families of α-helical barrel proteins with central channels that bind small molecules. Computational designs are seeded by the sequences and structures of defined de novo oligomeric barrel-forming peptides, and adjacent helices are connected by loop building. For targets with antiparallel helices, short loops are sufficient. However, targets with parallel helices require longer connectors; namely, an outer layer of helix-turn-helix-turn-helix motifs that are packed onto the barrels. Throughout these computational pipelines, residues that define open states of the barrels are maintained. This minimizes sequence sampling, accelerating the design process. For each of six targets, just two to six synthetic genes are made for expression in Escherichia coli. On average, 70% of these genes express to give soluble monomeric proteins that are fully characterized, including high-resolution structures for most targets that match the design models with high accuracy.
© 2024. The Author(s).
Conflict of interest statement
R.P. is a South West Biosciences Collaborative Awards in Science and Engineering student supported by Rosa Biotech. D.A.S. is a cofounder and was an employee at Rosa Biotech from 2019 to 2024. D.N.W. is a cofounder and director of Rosa Biotech. All other authors have no competing interests.
Figures