

Enterovirus 3A protein disrupts endoplasmic reticulum homeostasis through interaction with GBF1
- PMID: 38904364
- PMCID: PMC11265424
- DOI: 10.1128/jvi.00813-24
Enterovirus 3A protein disrupts endoplasmic reticulum homeostasis through interaction with GBF1
Abstract
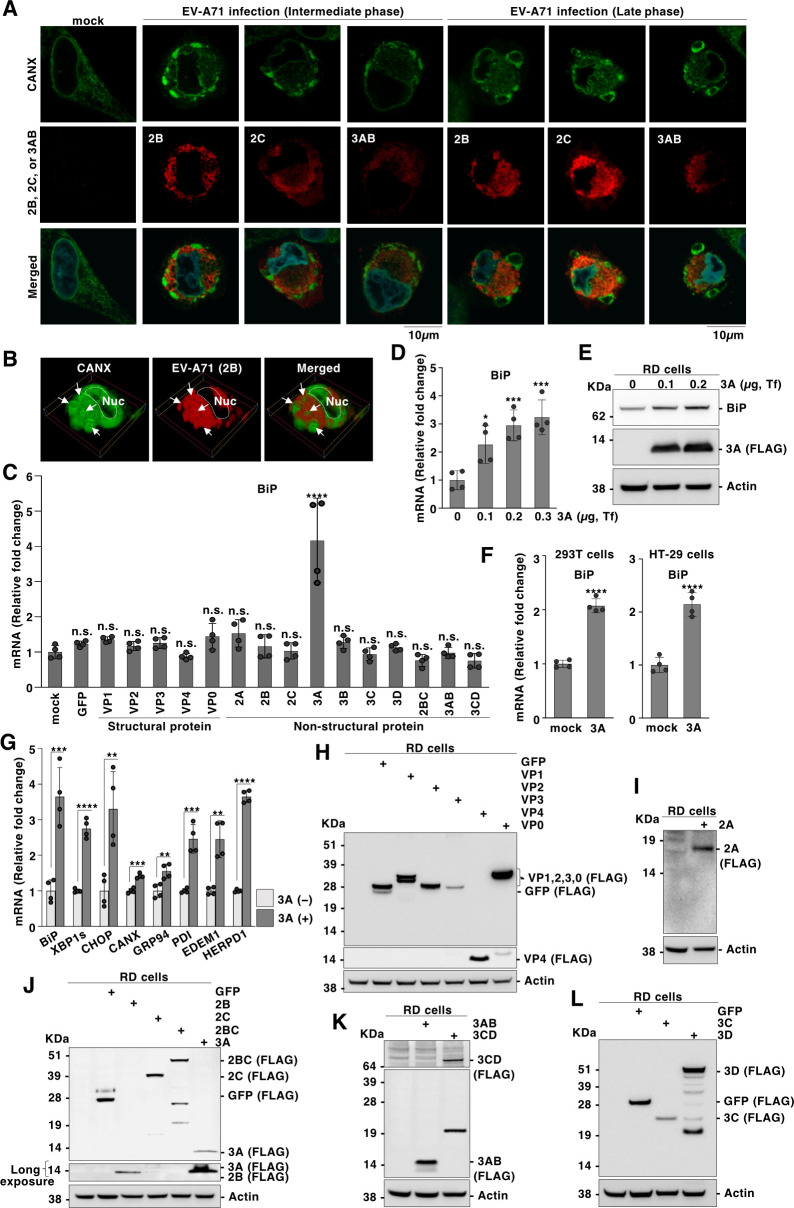
Enteroviruses are single-stranded, positive-sense RNA viruses causing endoplasmic reticulum (ER) stress to induce or modulate downstream signaling pathways known as the unfolded protein responses (UPR). However, viral and host factors involved in the UPR related to viral pathogenesis remain unclear. In the present study, we aimed to identify the major regulator of enterovirus-induced UPR and elucidate the underlying molecular mechanisms. We showed that host Golgi-specific brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF1), which supports enteroviruses replication, was a major regulator of the UPR caused by infection with enteroviruses. In addition, we found that severe UPR was induced by the expression of 3A proteins encoded in human pathogenic enteroviruses, such as enterovirus A71, coxsackievirus B3, poliovirus, and enterovirus D68. The N-terminal-conserved residues of 3A protein interact with the GBF1 and induce UPR through inhibition of ADP-ribosylation factor 1 (ARF1) activation via GBF1 sequestration. Remodeling and expansion of ER and accumulation of ER-resident proteins were observed in cells infected with enteroviruses. Finally, 3A induced apoptosis in cells infected with enteroviruses via activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK)/C/EBP homologous protein (CHOP) pathway of UPR. Pharmaceutical inhibition of PERK suppressed the cell death caused by infection with enteroviruses, suggesting the UPR pathway is a therapeutic target for treating diseases caused by infection with enteroviruses.IMPORTANCEInfection caused by several plus-stranded RNA viruses leads to dysregulated ER homeostasis in the host cells. The mechanisms underlying the disruption and impairment of ER homeostasis and its significance in pathogenesis upon enteroviral infection remain unclear. Our findings suggested that the 3A protein encoded in human pathogenic enteroviruses disrupts ER homeostasis by interacting with GBF1, a major regulator of UPR. Enterovirus-mediated infections drive ER into pathogenic conditions, where ER-resident proteins are accumulated. Furthermore, in such scenarios, the PERK/CHOP signaling pathway induced by an unresolved imbalance of ER homeostasis essentially drives apoptosis. Therefore, elucidating the mechanisms underlying the virus-induced disruption of ER homeostasis might be a potential target to mitigate the pathogenesis of enteroviruses.
Keywords: ER stress; GBF1; PERK; apoptosis; enterovirus; unfolded protein response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures











References
MeSH terms
Substances
Grants and funding
- JP22K15479/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- JP22K06625/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- JP22fk0108627j0601/Japan Agency for Medical Research and Development (AMED)
- SENSHIN Medical Research Foundation (SMRF)
- JPMJMS2025/MEXT | Japan Science and Technology Agency (JST)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
