

Retrograde adenosine/A2A receptor signaling facilitates excitatory synaptic transmission and seizures
- PMID: 38905101
- PMCID: PMC11286346
- DOI: 10.1016/j.celrep.2024.114382
Retrograde adenosine/A2A receptor signaling facilitates excitatory synaptic transmission and seizures
Abstract
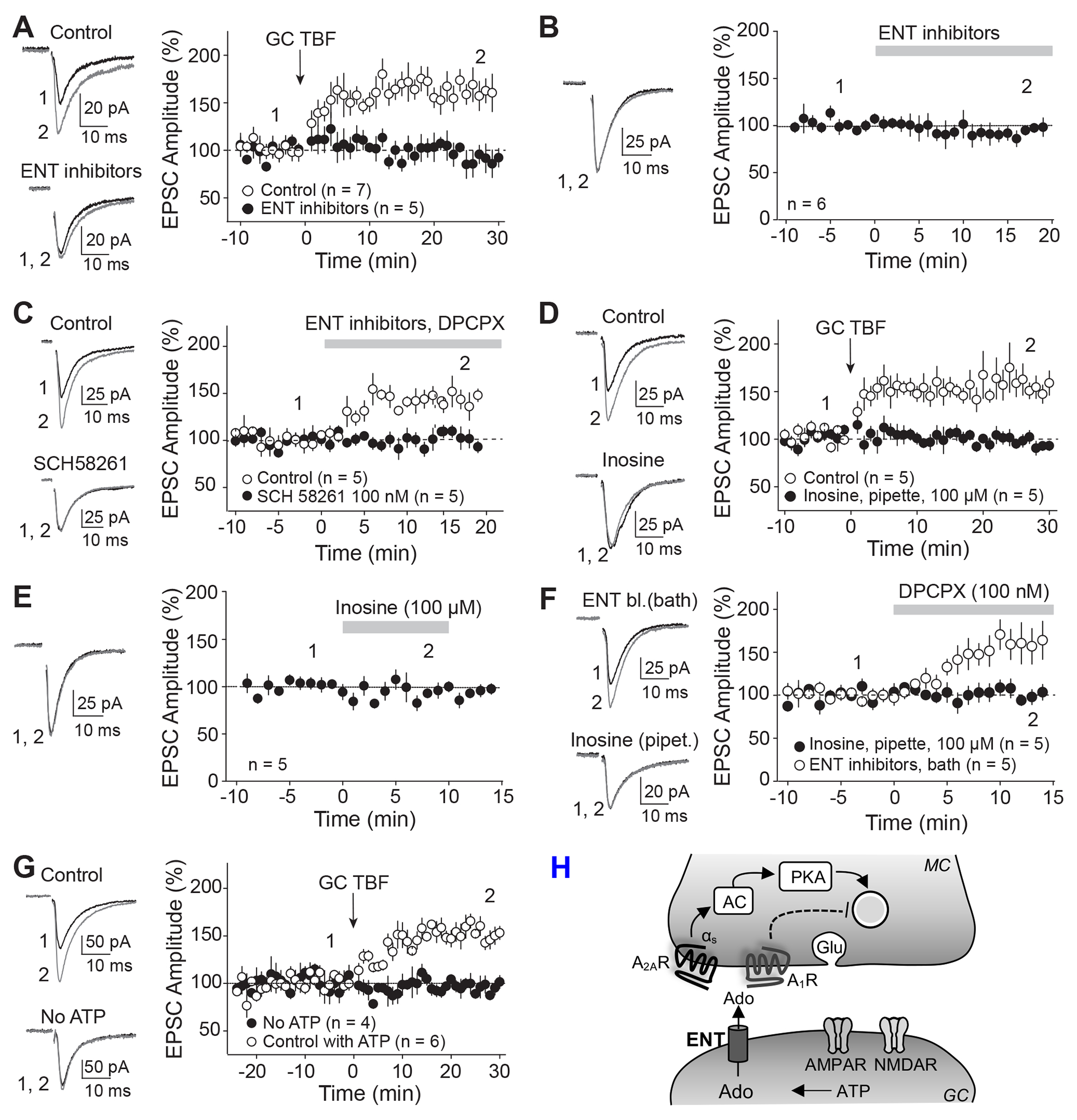
Retrograde signaling at the synapse is a fundamental way by which neurons communicate and neuronal circuit function is fine-tuned upon activity. While long-term changes in neurotransmitter release commonly rely on retrograde signaling, the mechanisms remain poorly understood. Here, we identified adenosine/A2A receptor (A2AR) as a retrograde signaling pathway underlying presynaptic long-term potentiation (LTP) at a hippocampal excitatory circuit critically involved in memory and epilepsy. Transient burst activity of a single dentate granule cell induced LTP of mossy cell synaptic inputs, a BDNF/TrkB-dependent form of plasticity that facilitates seizures. Postsynaptic TrkB activation released adenosine from granule cells, uncovering a non-conventional BDNF/TrkB signaling mechanism. Moreover, presynaptic A2ARs were necessary and sufficient for LTP. Lastly, seizure induction released adenosine in a TrkB-dependent manner, while removing A2ARs or TrkB from the dentate gyrus had anti-convulsant effects. By mediating presynaptic LTP, adenosine/A2AR retrograde signaling may modulate dentate gyrus-dependent learning and promote epileptic activity.
Keywords: BDNF; CP: Neuroscience; LTP; PKA; TrkB; dentate gyrus; epilepsy; hippocampus; mossy cell; presynaptic; retrograde signaling.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






Comment in
-
Adenosine Makes a Scene.Epilepsy Curr. 2025 Apr 16;25(3):198-200. doi: 10.1177/15357597251323126. eCollection 2025 May-Jun. Epilepsy Curr. 2025. PMID: 40256115 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
