

A multi-kingdom collection of 33,804 reference genomes for the human vaginal microbiome
- PMID: 38907008
- PMCID: PMC11306104
- DOI: 10.1038/s41564-024-01751-5
A multi-kingdom collection of 33,804 reference genomes for the human vaginal microbiome
Abstract
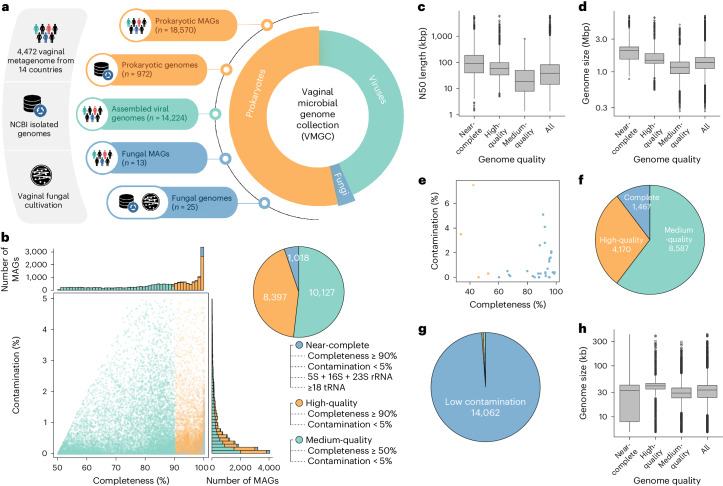
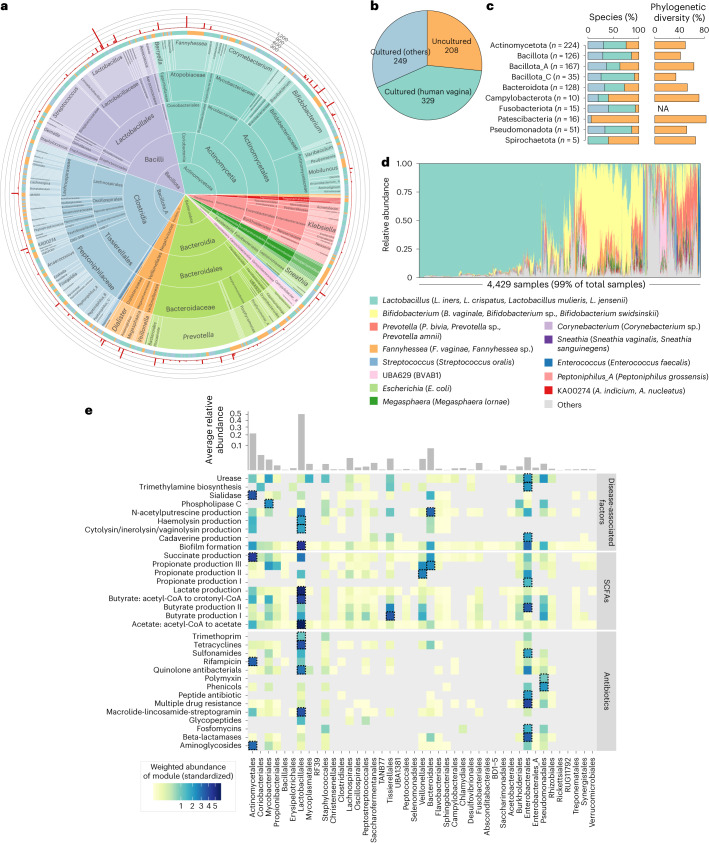
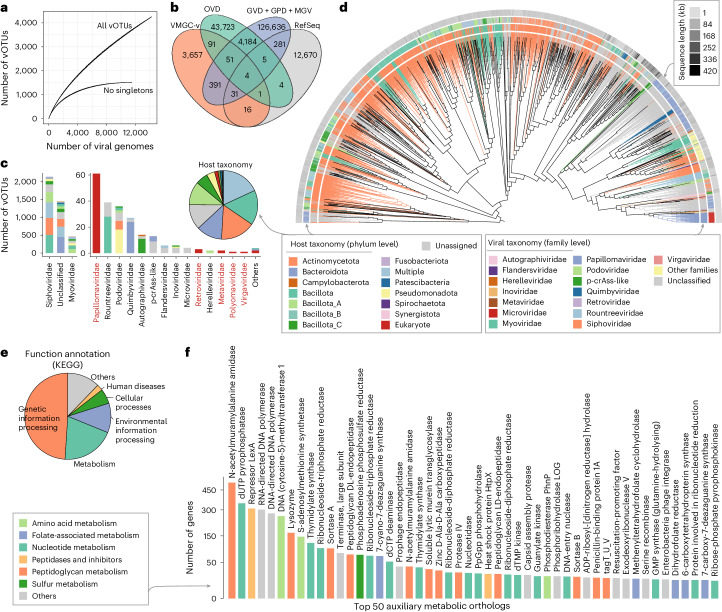
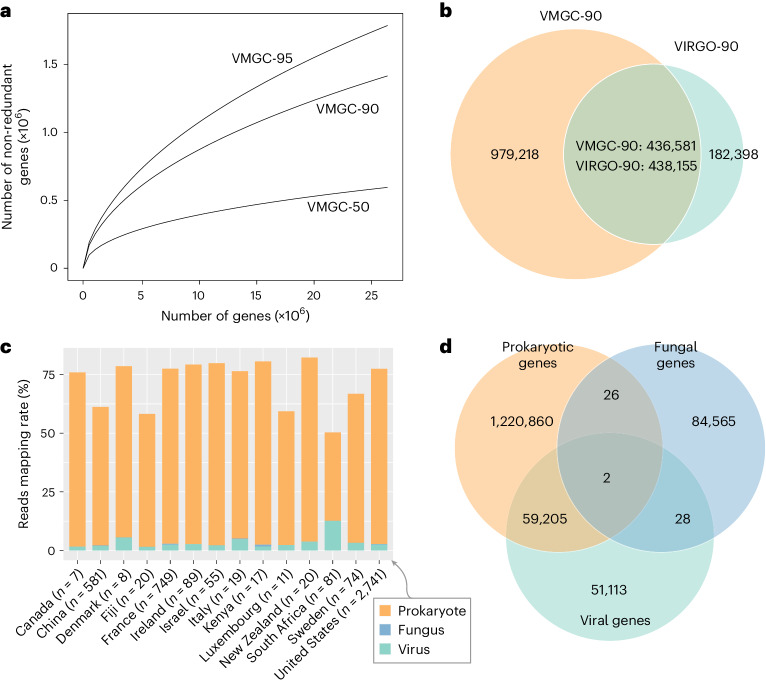
The human vagina harbours diverse microorganisms-bacteria, viruses and fungi-with profound implications for women's health. Genome-level analysis of the vaginal microbiome across multiple kingdoms remains limited. Here we utilize metagenomic sequencing data and fungal cultivation to establish the Vaginal Microbial Genome Collection (VMGC), comprising 33,804 microbial genomes spanning 786 prokaryotic species, 11 fungal species and 4,263 viral operational taxonomic units. Notably, over 25% of prokaryotic species and 85% of viral operational taxonomic units remain uncultured. This collection significantly enriches genomic diversity, especially for prevalent vaginal pathogens such as BVAB1 (an uncultured bacterial vaginosis-associated bacterium) and Amygdalobacter spp. (BVAB2 and related species). Leveraging VMGC, we characterize functional traits of prokaryotes, notably Saccharofermentanales (an underexplored yet prevalent order), along with prokaryotic and eukaryotic viruses, offering insights into their niche adaptation and potential roles in the vagina. VMGC serves as a valuable resource for studying vaginal microbiota and its impact on vaginal health.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
