

Ferroptosis in health and disease
- PMID: 38908072
- PMCID: PMC11253697
- DOI: 10.1016/j.redox.2024.103211
Ferroptosis in health and disease
Abstract
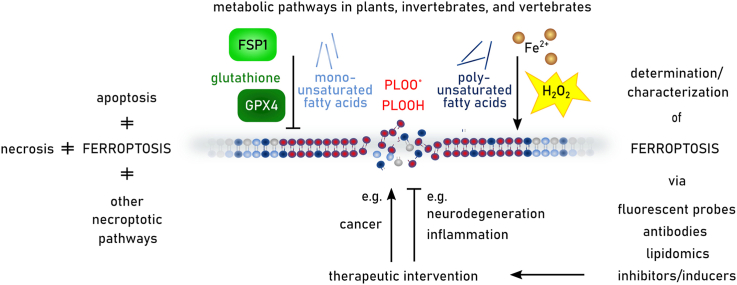
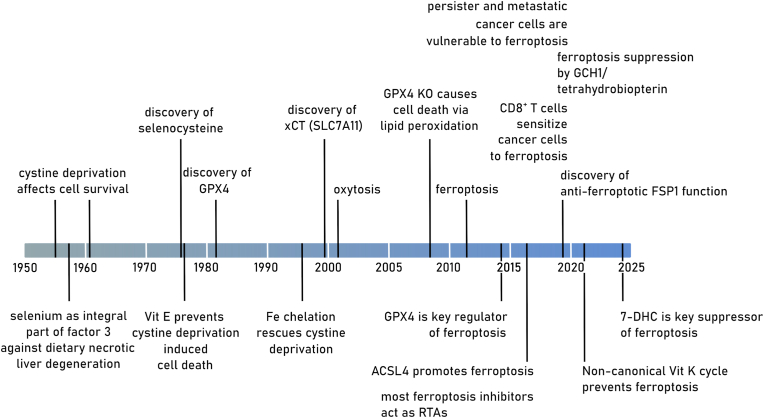
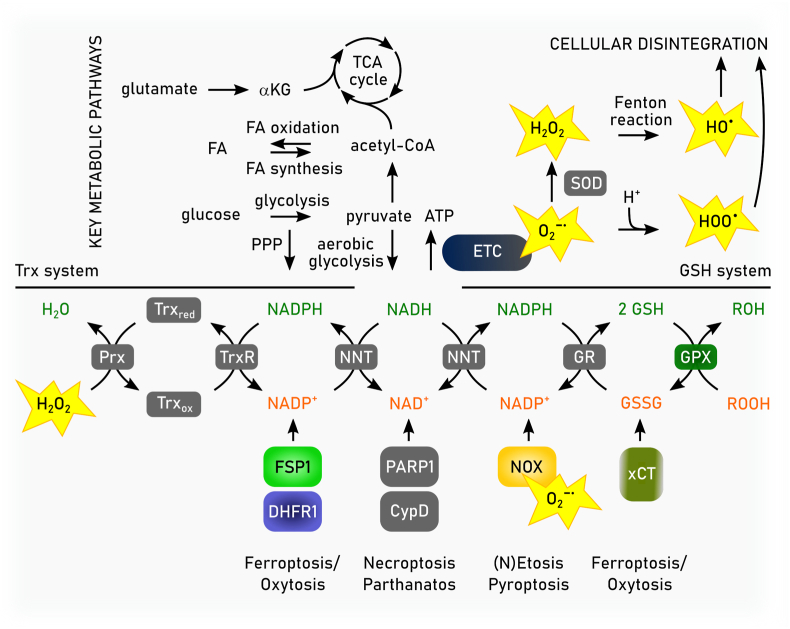
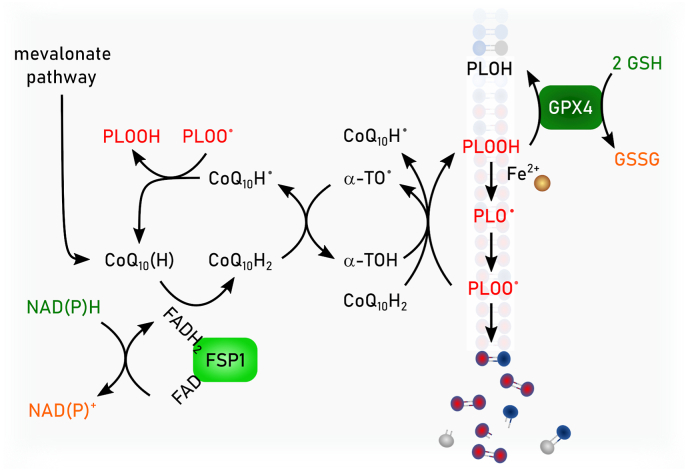
Ferroptosis is a pervasive non-apoptotic form of cell death highly relevant in various degenerative diseases and malignancies. The hallmark of ferroptosis is uncontrolled and overwhelming peroxidation of polyunsaturated fatty acids contained in membrane phospholipids, which eventually leads to rupture of the plasma membrane. Ferroptosis is unique in that it is essentially a spontaneous, uncatalyzed chemical process based on perturbed iron and redox homeostasis contributing to the cell death process, but that it is nonetheless modulated by many metabolic nodes that impinge on the cells' susceptibility to ferroptosis. Among the various nodes affecting ferroptosis sensitivity, several have emerged as promising candidates for pharmacological intervention, rendering ferroptosis-related proteins attractive targets for the treatment of numerous currently incurable diseases. Herein, the current members of a Germany-wide research consortium focusing on ferroptosis research, as well as key external experts in ferroptosis who have made seminal contributions to this rapidly growing and exciting field of research, have gathered to provide a comprehensive, state-of-the-art review on ferroptosis. Specific topics include: basic mechanisms, in vivo relevance, specialized methodologies, chemical and pharmacological tools, and the potential contribution of ferroptosis to disease etiopathology and progression. We hope that this article will not only provide established scientists and newcomers to the field with an overview of the multiple facets of ferroptosis, but also encourage additional efforts to characterize further molecular pathways modulating ferroptosis, with the ultimate goal to develop novel pharmacotherapies to tackle the various diseases associated with - or caused by - ferroptosis.
Keywords: Cancer; Cell death; Iron; Ischemia/reperfusion; Lipid peroxidation; Neurodegeneration.
Copyright © 2024 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: SJD is a co-founder of Prothegen, Inc. and holds patents related to ferroptosis. XJ is an inventor on patents related to autophagy and cell death and holds equity of and consults for Exarta Therapeutics and Lime Therapeutics. JAO is a member of the scientific advisory board for Vicinitas Therapeutics and has patent applications related to ferroptosis. TP reports grants from Dracen Pharmaceuticals, Kymera Therapeutics, Bristol-Myers Squibb, Agios Pharmaceuticals, personal fees from Vividion Therapeutics, Tohoku University, and personal fees from Faeth Therapeutics outside the submitted work; in addition, TP has a patent for US-20210361603-A1 pending and a patent for US-20210085763-A1 pending. BRS is an inventor on patents and patent applications involving ferroptosis, holds equity in and serves as a consultant to Exarta Therapeutics, and ProJenX Inc, holds equity in Sonata Therapeutics, and serves as a consultant to Weatherwax Biotechnologies Corporation and Akin Gump Strauss Hauer & Feld LLP. TVB holds patents related to ferroptosis inhibitors. CWil is a consultant for Odyssey Therapeutics and Orphagen Pharmaceuticals. YZ is a consultant for Keen Therapeutics. BP and MC hold patents for some of the compounds described herein, and is co-founder and shareholder of ROSCUE Therapeutics.
Figures
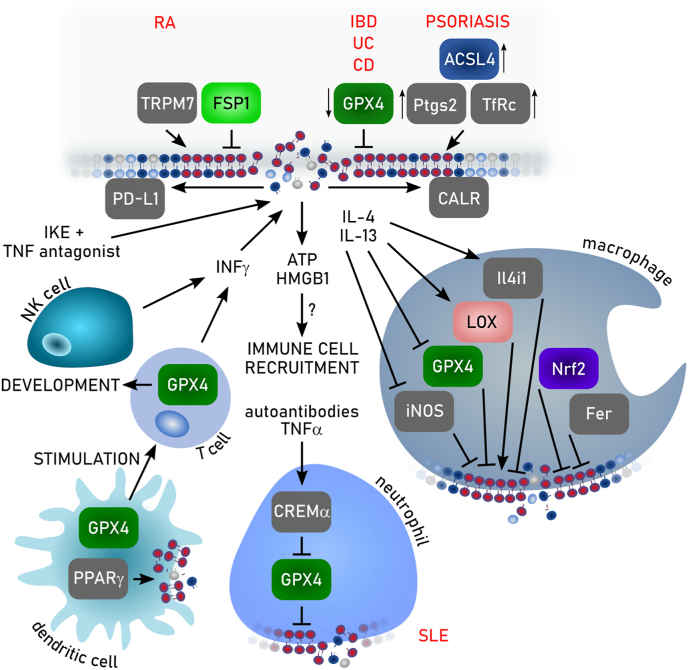
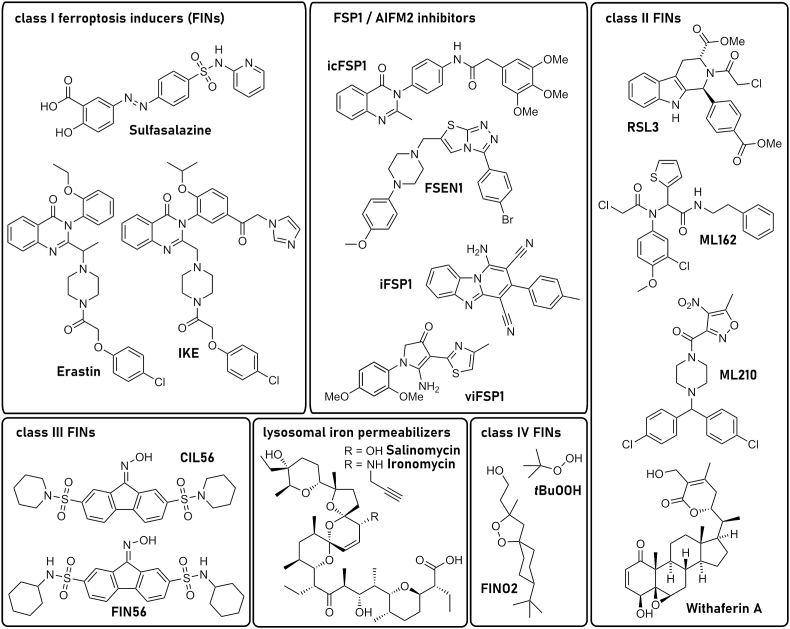
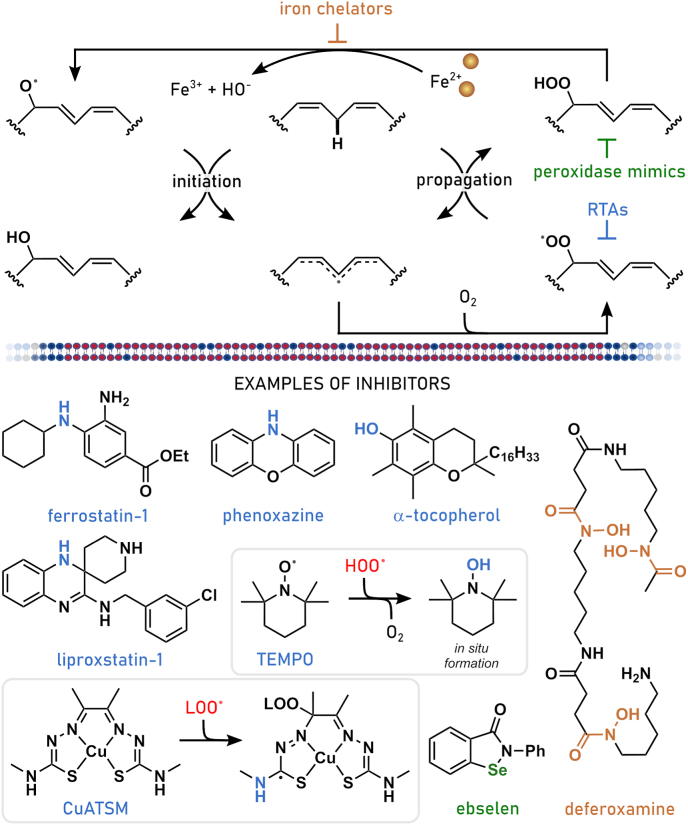
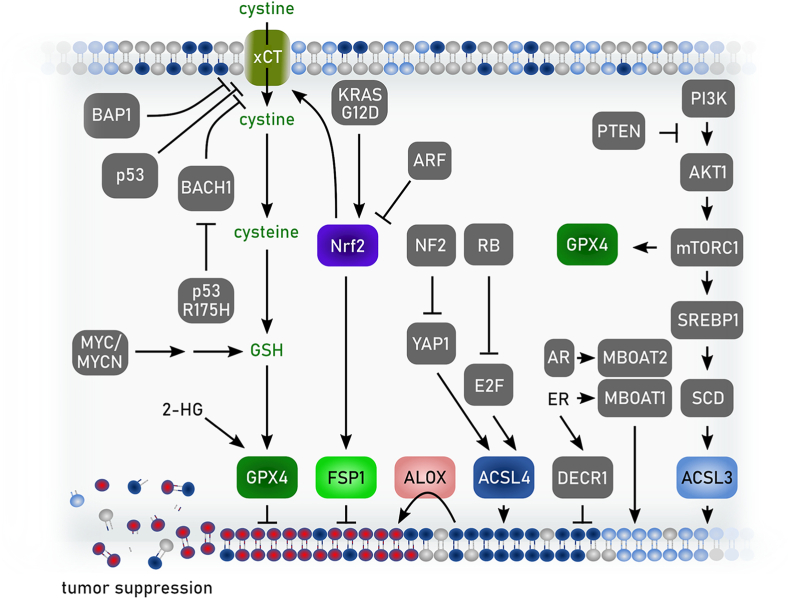
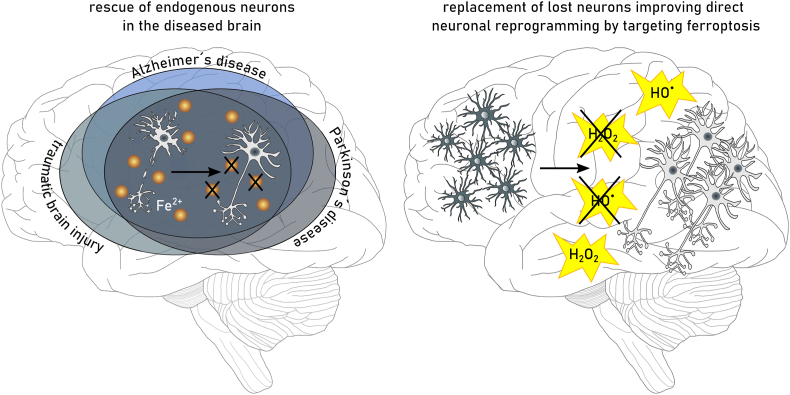
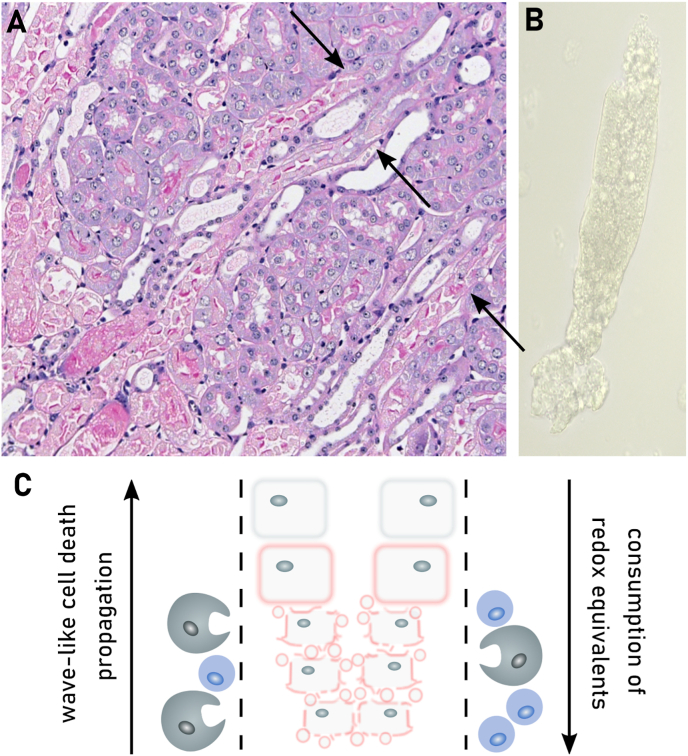
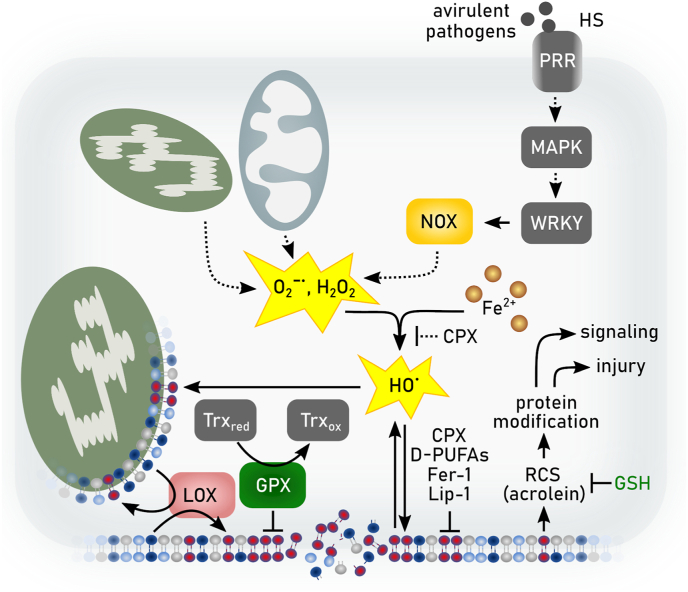
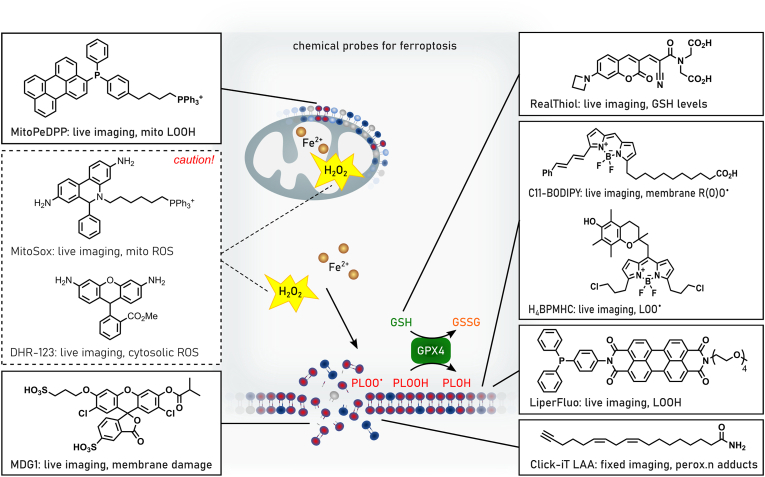
References
-
- Wiernicki B., Dubois H., Tyurina Y.Y., Hassannia B., Bayir H., Kagan V.E., Vandenabeele P., Wullaert A., Vanden Berghe T. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020;11:922. doi: 10.1038/s41419-020-03118-0. - DOI - PMC - PubMed
-
- Zilka O., Shah R., Li B., Friedmann Angeli J.P., Griesser M., Conrad M., Pratt D.A. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. - DOI - PMC - PubMed
-
- Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., 3rd, Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA165065/CA/NCI NIH HHS/United States
- R01 CA254970/CA/NCI NIH HHS/United States
- U01 AI156924/AI/NIAID NIH HHS/United States
- R01 NS076511/NS/NINDS NIH HHS/United States
- R01 AI145406/AI/NIAID NIH HHS/United States
- R01 NS061817/NS/NINDS NIH HHS/United States
- R01 CA243142/CA/NCI NIH HHS/United States
- P01 HL114453/HL/NHLBI NIH HHS/United States
- R35 CA253059/CA/NCI NIH HHS/United States
- R01 GM112948/GM/NIGMS NIH HHS/United States
- R01 CA258390/CA/NCI NIH HHS/United States
- R37 NS061817/NS/NINDS NIH HHS/United States
- R01 CA266342/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U01 AI156923/AI/NIAID NIH HHS/United States
