

Demultiplexing of single-cell RNA-sequencing data using interindividual variation in gene expression
- PMID: 38911824
- PMCID: PMC11193101
- DOI: 10.1093/bioadv/vbae085
Demultiplexing of single-cell RNA-sequencing data using interindividual variation in gene expression
Abstract
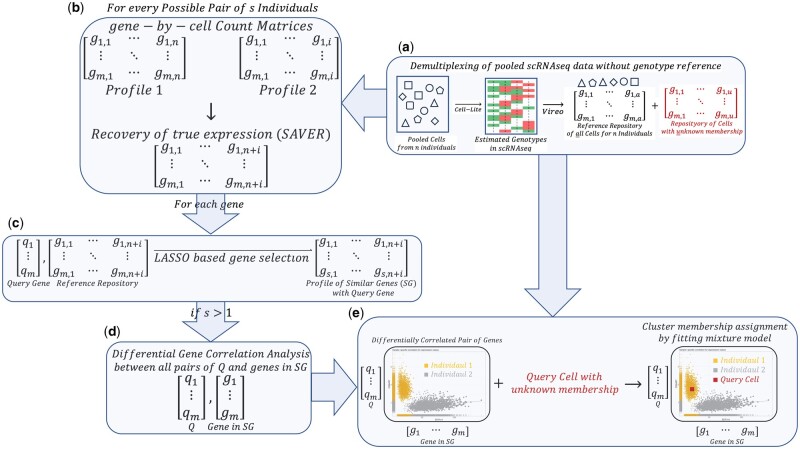
Motivation: Pooled designs for single-cell RNA sequencing, where many cells from distinct samples are processed jointly, offer increased throughput and reduced batch variation. This study describes expression-aware demultiplexing (EAD), a computational method that employs differential co-expression patterns between individuals to demultiplex pooled samples without any extra experimental steps.
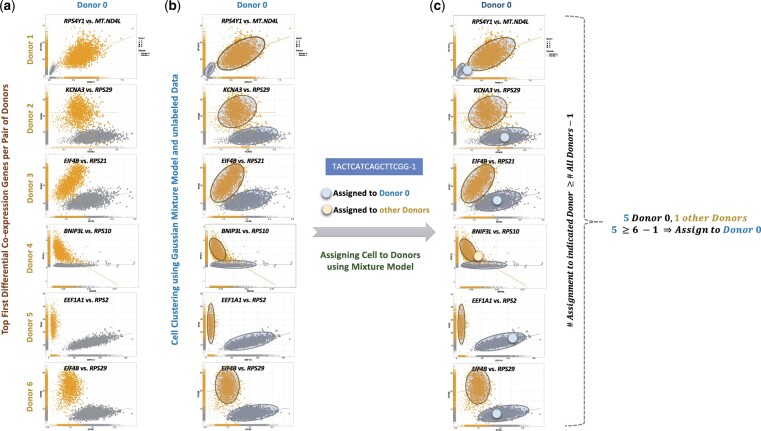
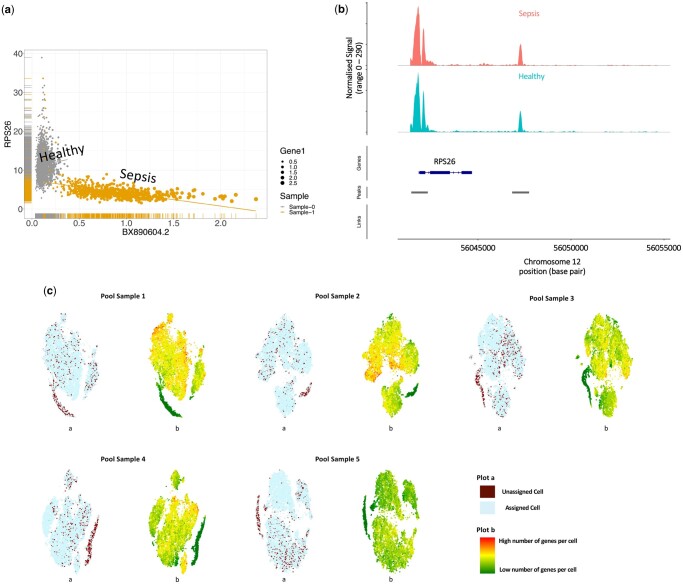
Results: We use synthetic sample pools and show that the top interindividual differentially co-expressed genes provide a distinct cluster of cells per individual, significantly enriching the regulation of metabolism. Our application of EAD to samples of six isogenic inbred mice demonstrated that controlling genetic and environmental effects can solve interindividual variations related to metabolic pathways. We utilized 30 samples from both sepsis and healthy individuals in six batches to assess the performance of classification approaches. The results indicate that combining genetic and EAD results can enhance the accuracy of assignments (Min. 0.94, Mean 0.98, Max. 1). The results were enhanced by an average of 1.4% when EAD and barcoding techniques were combined (Min. 1.25%, Median 1.33%, Max. 1.74%). Furthermore, we demonstrate that interindividual differential co-expression analysis within the same cell type can be used to identify cells from the same donor in different activation states. By analysing single-nuclei transcriptome profiles from the brain, we demonstrate that our method can be applied to nonimmune cells.
Availability and implementation: EAD workflow is available at https://isarnassiri.github.io/scDIV/ as an R package called scDIV (acronym for single-cell RNA-sequencing data demultiplexing using interindividual variations).
© Crown copyright 2024.
Conflict of interest statement
None declared.
Figures






References
-
- Almeida A, Loy A, Hofmann H.. ggplot2 compatible quantile-quantile plots in R. R J 2018;10:248–61.
-
- Badia-I-Mompel P, Wessels L, Müller-Dott S. et al. Gene regulatory network inference in the era of single-cell multi-omics. Nat Rev Genet 2023;24:739–54. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
