

Histone demethylase JARID1C/KDM5C regulates Th17 cells by increasing IL-6 expression in diabetic plasmacytoid dendritic cells
- PMID: 38912581
- PMCID: PMC11383169
- DOI: 10.1172/jci.insight.172959
Histone demethylase JARID1C/KDM5C regulates Th17 cells by increasing IL-6 expression in diabetic plasmacytoid dendritic cells
Abstract
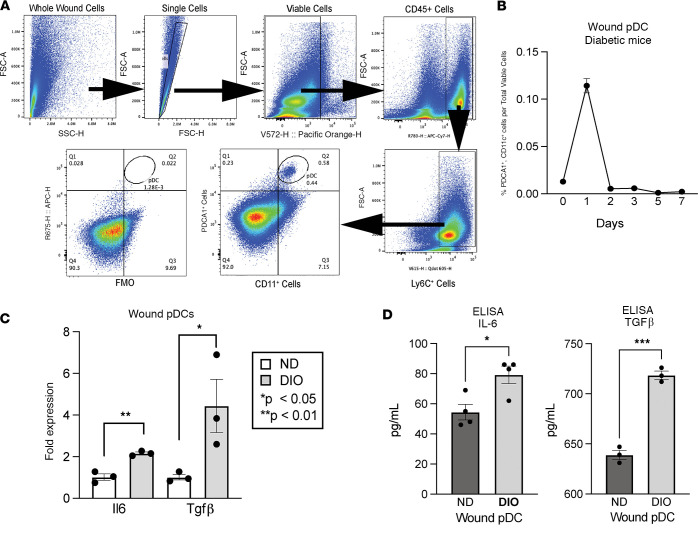
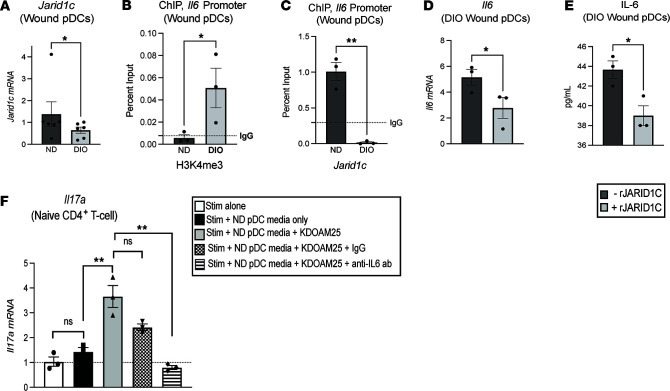
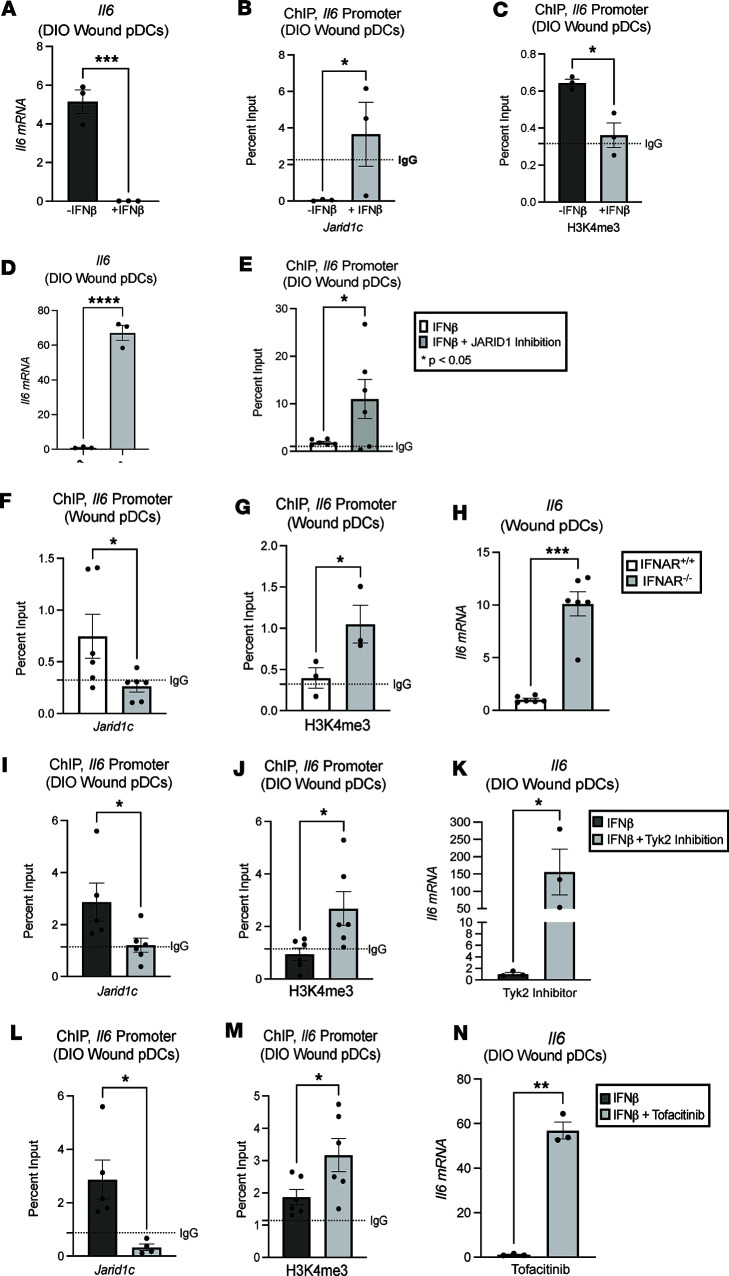
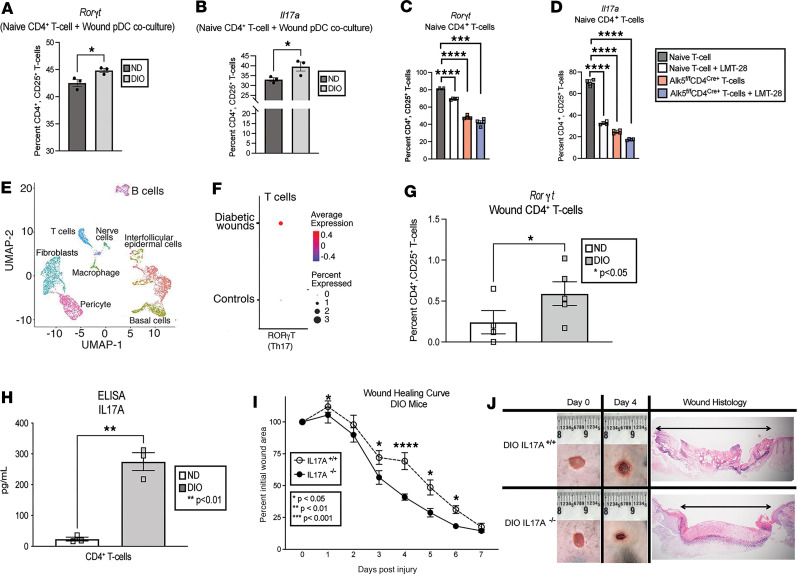
Plasmacytoid dendritic cells (pDCs) are first responders to tissue injury, where they prime naive T cells. The role of pDCs in physiologic wound repair has been examined, but little is known about pDCs in diabetic wound tissue and their interactions with naive CD4+ T cells. Diabetic wounds are characterized by increased levels of inflammatory IL-17A cytokine, partly due to increased Th17 CD4+ cells. This increased IL-17A cytokine, in excess, impairs tissue repair. Here, using human tissue and murine wound healing models, we found that diabetic wound pDCs produced excess IL-6 and TGF-β and that these cytokines skewed naive CD4+ T cells toward a Th17 inflammatory phenotype following cutaneous injury. Further, we identified that increased IL-6 cytokine production by diabetic wound pDCs is regulated by a histone demethylase, Jumonji AT-rich interactive domain 1C histone demethylase (JARID1C). Decreased JARID1C increased IL-6 transcription in diabetic pDCs, and this process was regulated upstream by an IFN-I/TYK2/JAK1,3 signaling pathway. When inhibited in nondiabetic wound pDCs, JARID1C skewed naive CD4+ T cells toward a Th17 phenotype and increased IL-17A production. Together, this suggests that diabetic wound pDCs are epigenetically altered to increase IL-6 expression that then affects T cell phenotype. These findings identify a therapeutically manipulable pathway in diabetic wounds.
Keywords: Adaptive immunity; Dendritic cells; Epigenetics; Immunology; Inflammation.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
- R00 DK133828/DK/NIDDK NIH HHS/United States
- R01 HL137919/HL/NHLBI NIH HHS/United States
- R01 DK127531/DK/NIDDK NIH HHS/United States
- R01 HL156274/HL/NHLBI NIH HHS/United States
- R35 HL167143/HL/NHLBI NIH HHS/United States
- K99 DK133828/DK/NIDDK NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- R01 DK124290/DK/NIDDK NIH HHS/United States
- T32 AI007413/AI/NIAID NIH HHS/United States
- R35 HL144481/HL/NHLBI NIH HHS/United States
- R01 AR079863/AR/NIAMS NIH HHS/United States
- R01 HL156275/HL/NHLBI NIH HHS/United States
- F32 DK126471/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
