

Detecting Small Cell Transformation in Patients with Advanced EGFR Mutant Lung Adenocarcinoma through Epigenomic cfDNA Profiling
- PMID: 38912901
- PMCID: PMC11369616
- DOI: 10.1158/1078-0432.CCR-24-0466
Detecting Small Cell Transformation in Patients with Advanced EGFR Mutant Lung Adenocarcinoma through Epigenomic cfDNA Profiling
Abstract
Purpose: Histologic transformation to small cell lung cancer (SCLC) is a mechanism of treatment resistance in patients with advanced oncogene-driven lung adenocarcinoma (LUAD) that currently requires histologic review for diagnosis. Herein, we sought to develop an epigenomic cell-free DNA (cfDNA)-based approach to noninvasively detect small cell transformation in patients with EGFR mutant (EGFRm) LUAD.
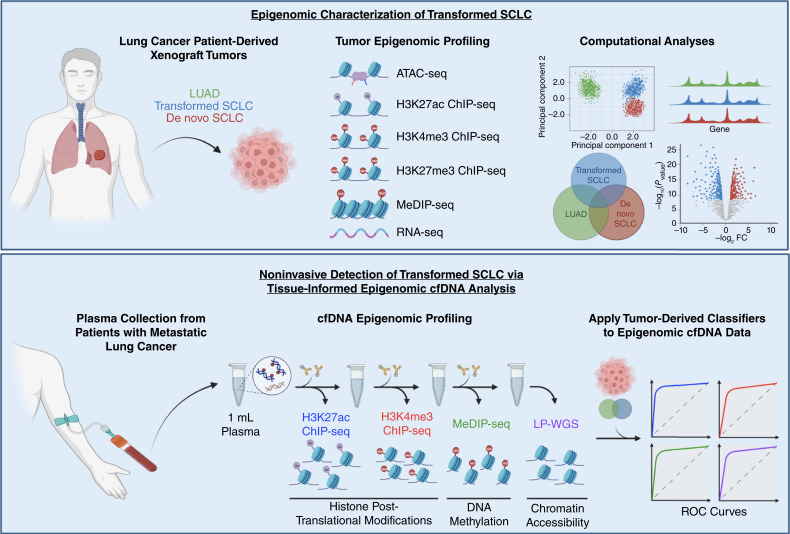
Experimental design: To characterize the epigenomic landscape of transformed (t)SCLC relative to LUAD and de novo SCLC, we performed chromatin immunoprecipitation sequencing (ChIP-seq) to profile the histone modifications H3K27ac, H3K4me3, and H3K27me3; methylated DNA immunoprecipitation sequencing (MeDIP-seq); assay for transposase-accessible chromatin sequencing; and RNA sequencing on 26 lung cancer patient-derived xenograft (PDX) tumors. We then generated and analyzed H3K27ac ChIP-seq, MeDIP-seq, and whole genome sequencing cfDNA data from 1 mL aliquots of plasma from patients with EGFRm LUAD with or without tSCLC.
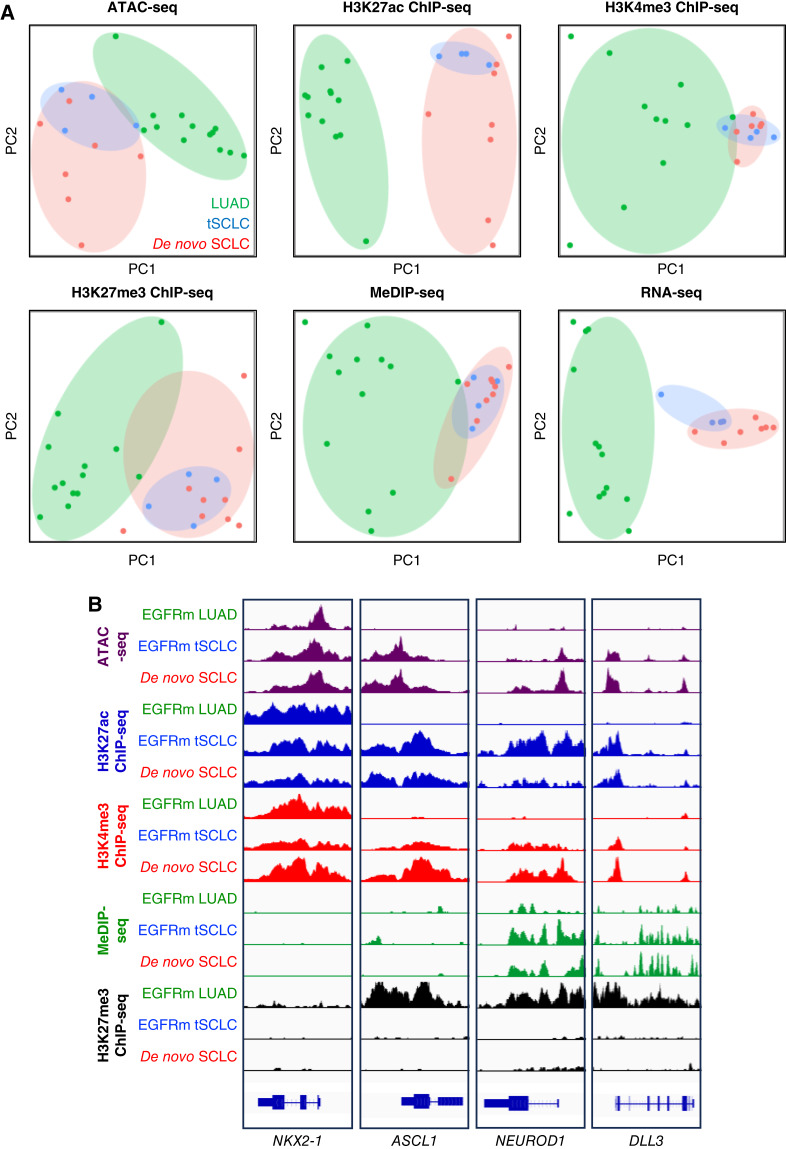
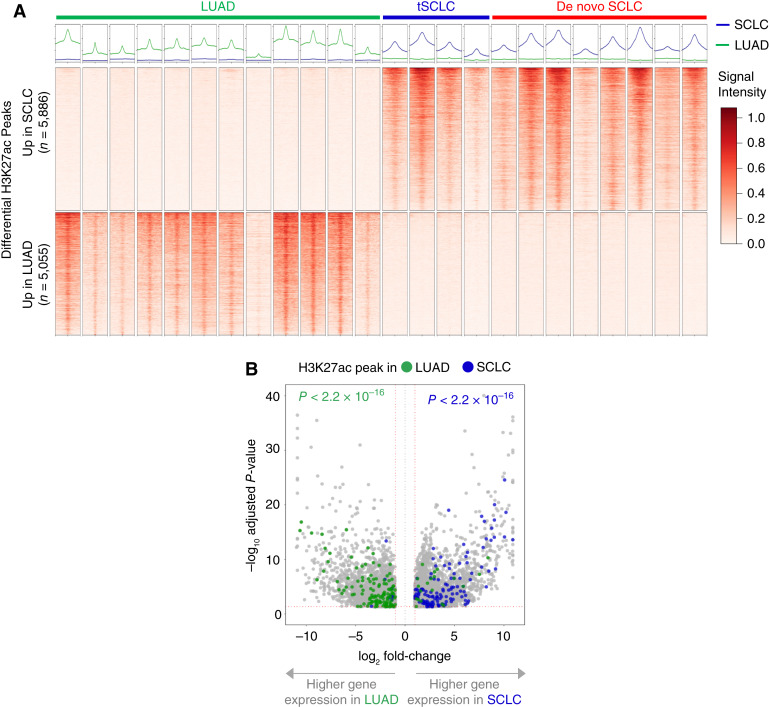
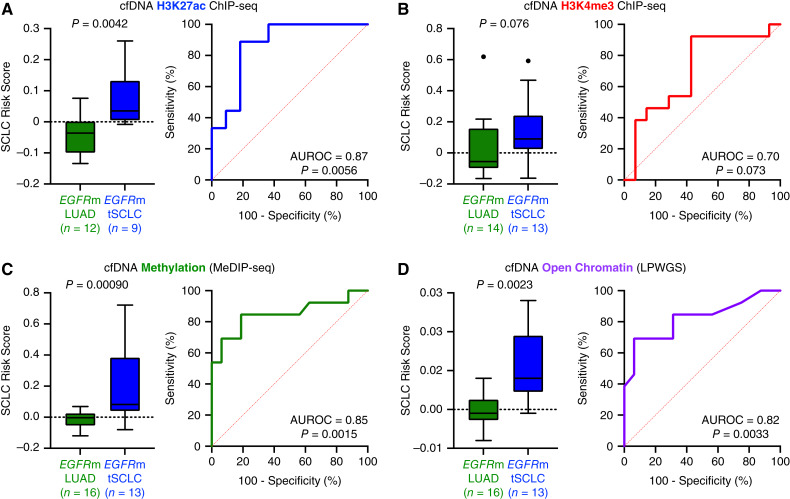
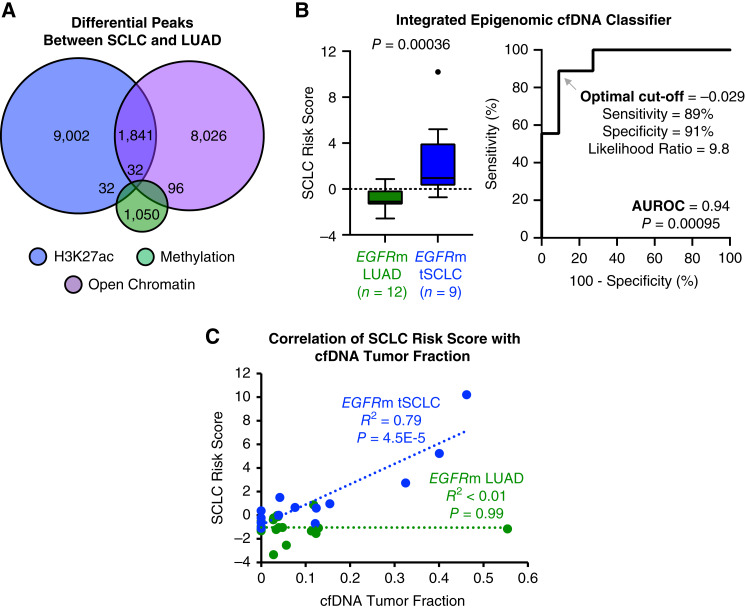
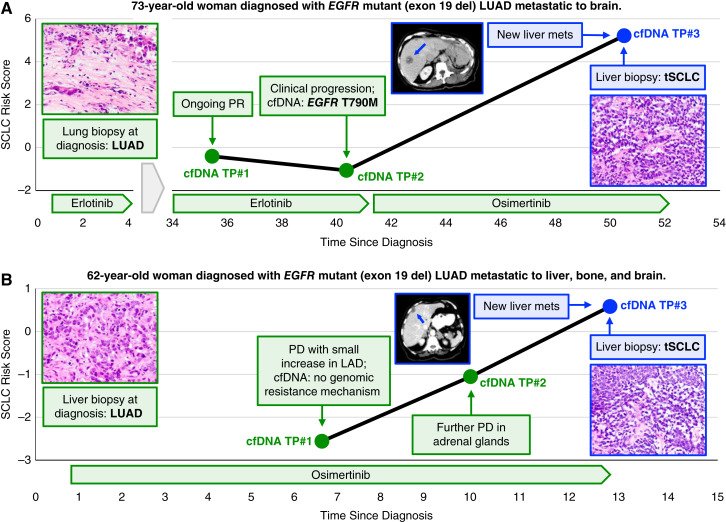
Results: Analysis of 126 epigenomic libraries from the lung cancer PDXs revealed widespread epigenomic reprogramming between LUAD and tSCLC, with a large number of differential H3K27ac (n = 24,424), DNA methylation (n = 3,298), and chromatin accessibility (n = 16,352) sites between the two histologies. Tumor-informed analysis of each of these three epigenomic features in cfDNA resulted in accurate noninvasive discrimination between patients with EGFRm LUAD versus tSCLC [area under the receiver operating characteristic curve (AUROC) = 0.82-0.87]. A multianalyte cfDNA-based classifier integrating these three epigenomic features discriminated between EGFRm LUAD versus tSCLC with an AUROC of 0.94.
Conclusions: These data demonstrate the feasibility of detecting small cell transformation in patients with EGFRm LUAD through epigenomic cfDNA profiling of 1 mL of patient plasma.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
Y.P. Hung reports honoraria from Elsevier and American Society of Clinical Pathology on textbook writing and continuing medical education–related activity, both of which are unrelated to this study. N.R. Mahadevan reports stock ownership in AstraZeneca and Roche. D.A. Barbie reports personal fees from Qiagen/N of One and other support from Xsphera Biosciences outside the submitted work. Z. Piotrowska reports grants from NIH during the conduct of the study. Z. Piotrowska also reports personal fees from Eli Lilly, Boehringer Ingelheim, Bayer, Sanofi, C4 Therapeutics, and Taiho Pharmaceuticals; grants, personal fees, and other support from Janssen and AstraZeneca; grants and personal fees from Takeda, Cullinan Oncology, Daiichi Sankyo, and Blueprint Medicines; grants from Novartis, Spectrum Pharmaceuticals, AbbVie, GlaxoSmithKline/Tesaro, and Phanes Therapeutics; and grants and other support from Genentech/Roche outside the submitted work. T.K. Choueiri reports personal fees and other support from Precede Bio during the conduct of the study, as well as grants and other support from Precede Bio outside the submitted work; in addition, T.K. Choueiri has a patent for Precede Bio with royalties paid. T.K. Choueiri also reports institutional and/or personal, paid and/or unpaid support for research, advisory boards, consultancy, and/or honoraria past 5 years, ongoing or not, from Alkermes, Arcus Bio, AstraZeneca, Aravive, Aveo, Bayer, Bristol Myers Squibb, Calithera, Circle Pharma, Deciphera Pharmaceuticals, Eisai, EMD Serono, Exelixis, GlaxoSmithKline, Gilead, HiberCell, IQVA, Infinity, Ipsen, Janssen, Kanaph, Lilly, Merck, Nikang, Neomorph, Nuscan/Precede Bio, Novartis, Oncohost, Pfizer, Roche, Sanofi/Aventis, Scholar Rock, Surface Oncology, Takeda, Tempest, Up-To-Date, CME events (Peerview, OncLive, MJH, CCO and others), outside the submitted work; institutional patents filed on molecular alterations and immunotherapy response/toxicity, and ctDNA; equity from Tempest, Pionyr, Osel, Precede Bio, CureResponse, InnDura Therapeutics, and Primium; committees for NCCN, GU Steering Committee, ASCO (BOD 6-2024-), ESMO, ACCRU, and KidneyCan; medical writing and editorial assistance support may have been funded by communications companies in part; mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/foreign components; and the institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies and/or royalties potentially involved in research around the subject matter. T.K. Choueiri is supported in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE (2P50CA101942-16) and Program 5P30CA006516-56, the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, Pan Mass Challenge, Hinda and Arthur Marcus Fund, and Loker Pinard Funds for Kidney Cancer Research at DFCI. S.C. Baca reports personal fees and other support from Precede Biosciences outside the submitted work. A.N. Hata reports grants and personal fees from Amgen, Nuvalent, and Pfizer; grants from BridgeBio, Bristol-Myers Squibb, C4 Therapeutics, Eli Lilly, Novartis, and Scorpion Therapeutics; and personal fees from Engine Biosciences, Oncovalent, TigaTx, and Tolremo outside the submitted work. M.L. Freedman reports personal fees and other support from Precede Biosciences outside the submitted work; in addition, M.L. Freedman has a patent for 'Methods, kits and systems for determining the status of lung cancer and methods for treating lung cancer based on same' pending. J.E. Berchuck reports non-financial support and other support from Precede Biosciences during the conduct of the study. J.E. Berchuck also reports grants, personal fees, and non-financial support from Guardant Health; personal fees and other support from Genome Medical; and other support from Oncotect, TracerDx, and Musculo outside the submitted work. In addition, J.E. Berchuck has an institutional patent on methods to detect neuroendocrine prostate cancer through tissue-informed cell-free DNA methylation analysis issued, licensed, and with royalties paid from Precede Biosciences and an institutional patent on methods to detect small cell lung cancer through epigenomic cfDNA analysis pending. No disclosures were reported by the other authors.
Figures
References
-
- National Comprehensive Cancer Network Practice Guidelines in Oncology (NCCN Guidelines) . Non-small cell lung cancer version 1.2024. National Comprehensive Cancer Network. - PubMed
-
- Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 2018;378:113–25. - PubMed
-
- Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov 2018;8:1529–39. - PMC - PubMed
MeSH terms
Substances
Grants and funding
- Cutler Family Fund for Prevention and Early Detection
- H.L. Snyder Medical Research Foundation
- Damon Runyon Cancer Research Foundation (DRCRF)
- Claudia Adams Barr Program for Innovative Cancer Research
- W81XWH-21-1-0358/DOD Prostate Cancer Research Program (PCRP)
- LUNGSTRONG Foundation
- R01 CA137008/CA/NCI NIH HHS/United States
- Donahue Family Fund
- R01 CA251555/CA/NCI NIH HHS/United States
- Dana-Farber Cancer Institute Presidential Initiatives Fund
- R01CA137008/National Cancer Institute (NCI)
- Fund for Innovation in Cancer Informatics (ICI)
- W81XWH-21-1-0339/Congressionally Directed Medical Research Programs (CDMRP)
- K08 CA270077/CA/NCI NIH HHS/United States
- 2022A019104/American Cancer Society (ACS)
- R01CA262577/National Cancer Institute (NCI)
- Center Lung Cancer SPORE Research Development Award/Dana-Farber/Harvard Cancer Center (DF/HCC)
- The Dave Page Cancer Research Fund
- R01 CA262577/CA/NCI NIH HHS/United States
- K08CA270077/National Cancer Institute (NCI)
- W81XWH-20-1-0118/DOD Prostate Cancer Research Program (PCRP)
- Movember PCF Challenge Award/Prostate Cancer Foundation (PCF)
- Kure It Cancer Research Foundation
- Pan-Mass Challenge Team 3G
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
