

Pairing interacting protein sequences using masked language modeling
- PMID: 38913900
- PMCID: PMC11228504
- DOI: 10.1073/pnas.2311887121
Pairing interacting protein sequences using masked language modeling
Abstract
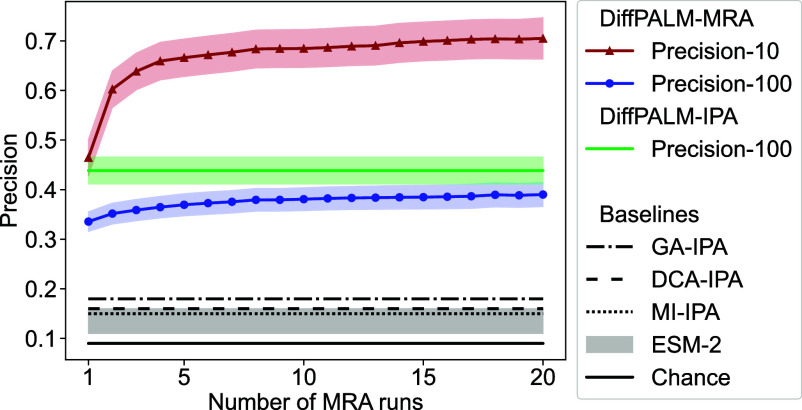
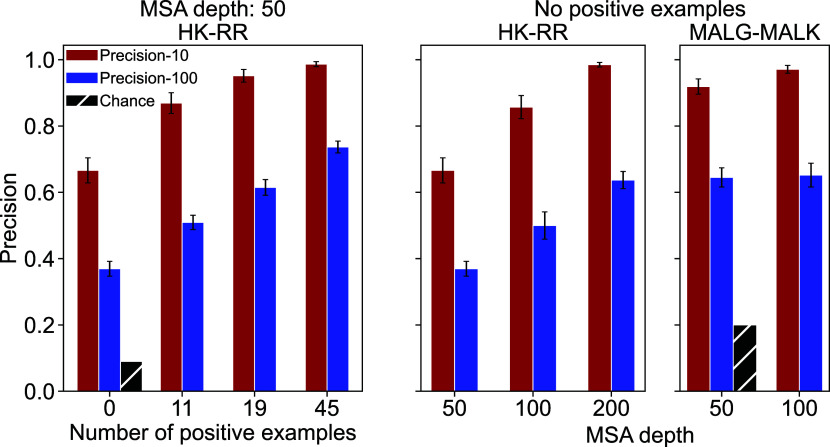
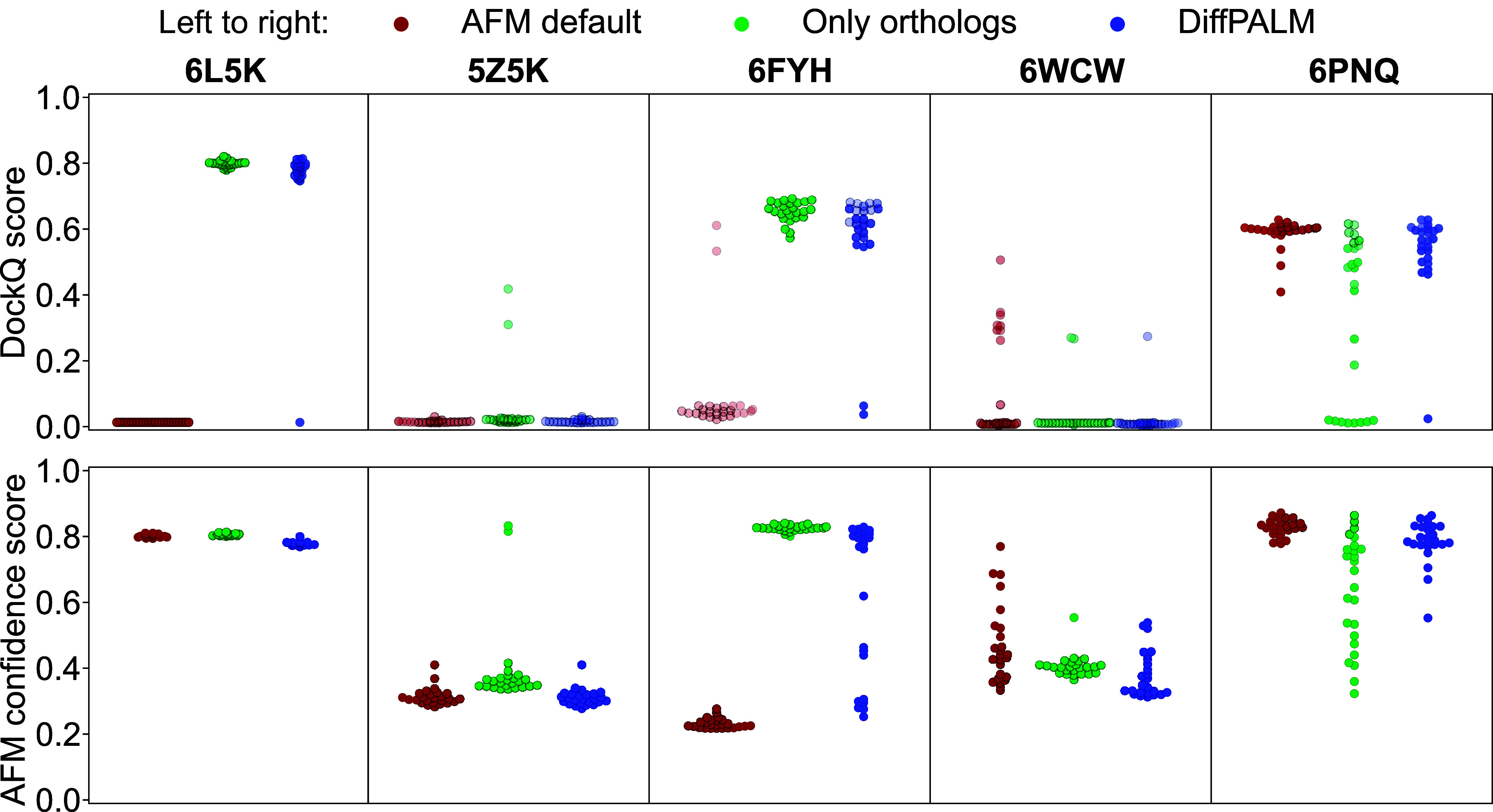
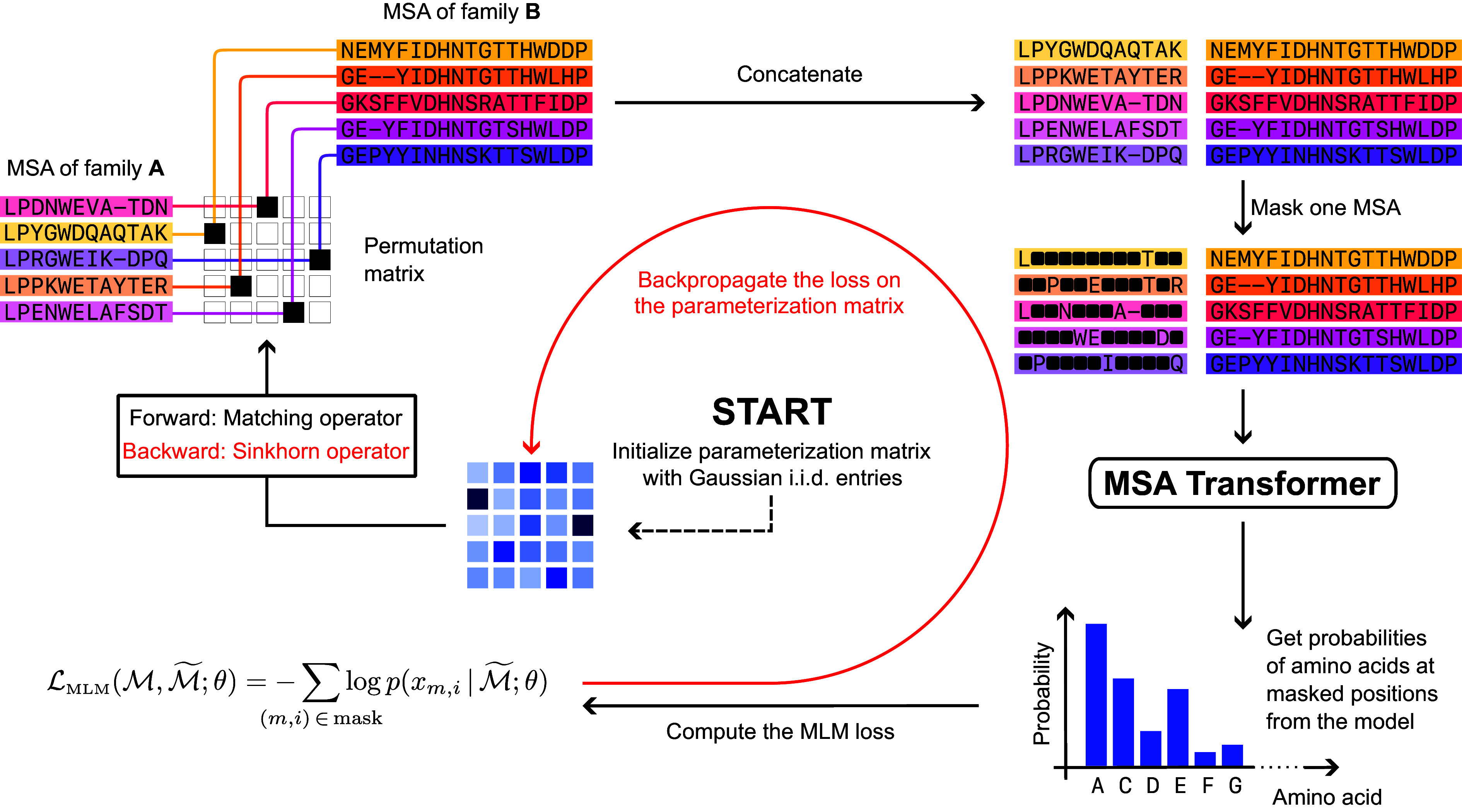
Predicting which proteins interact together from amino acid sequences is an important task. We develop a method to pair interacting protein sequences which leverages the power of protein language models trained on multiple sequence alignments (MSAs), such as MSA Transformer and the EvoFormer module of AlphaFold. We formulate the problem of pairing interacting partners among the paralogs of two protein families in a differentiable way. We introduce a method called Differentiable Pairing using Alignment-based Language Models (DiffPALM) that solves it by exploiting the ability of MSA Transformer to fill in masked amino acids in multiple sequence alignments using the surrounding context. MSA Transformer encodes coevolution between functionally or structurally coupled amino acids within protein chains. It also captures inter-chain coevolution, despite being trained on single-chain data. Relying on MSA Transformer without fine-tuning, DiffPALM outperforms existing coevolution-based pairing methods on difficult benchmarks of shallow multiple sequence alignments extracted from ubiquitous prokaryotic protein datasets. It also outperforms an alternative method based on a state-of-the-art protein language model trained on single sequences. Paired alignments of interacting protein sequences are a crucial ingredient of supervised deep learning methods to predict the three-dimensional structure of protein complexes. Starting from sequences paired by DiffPALM substantially improves the structure prediction of some eukaryotic protein complexes by AlphaFold-Multimer. It also achieves competitive performance with using orthology-based pairing.
Keywords: coevolution; machine learning; protein complex structure; protein language models; protein–protein interactions.
Conflict of interest statement
Competing interests statement:The authors declare no competing interest.
Figures
Similar articles
-
The Historical Evolution and Significance of Multiple Sequence Alignment in Molecular Structure and Function Prediction.Biomolecules. 2024 Nov 29;14(12):1531. doi: 10.3390/biom14121531. Biomolecules. 2024. PMID: 39766238 Free PMC article. Review.
-
DiffPaSS-high-performance differentiable pairing of protein sequences using soft scores.Bioinformatics. 2024 Dec 26;41(1):btae738. doi: 10.1093/bioinformatics/btae738. Bioinformatics. 2024. PMID: 39672677 Free PMC article.
-
Generative power of a protein language model trained on multiple sequence alignments.Elife. 2023 Feb 3;12:e79854. doi: 10.7554/eLife.79854. Elife. 2023. PMID: 36734516 Free PMC article.
-
Protein language models trained on multiple sequence alignments learn phylogenetic relationships.Nat Commun. 2022 Oct 22;13(1):6298. doi: 10.1038/s41467-022-34032-y. Nat Commun. 2022. PMID: 36273003 Free PMC article.
-
Recent advances in features generation for membrane protein sequences: From multiple sequence alignment to pre-trained language models.Proteomics. 2023 Dec;23(23-24):e2200494. doi: 10.1002/pmic.202200494. Epub 2023 Oct 20. Proteomics. 2023. PMID: 37863817 Review.
Cited by
-
The Historical Evolution and Significance of Multiple Sequence Alignment in Molecular Structure and Function Prediction.Biomolecules. 2024 Nov 29;14(12):1531. doi: 10.3390/biom14121531. Biomolecules. 2024. PMID: 39766238 Free PMC article. Review.
-
Machine learning meets physics: A two-way street.Proc Natl Acad Sci U S A. 2024 Jul 2;121(27):e2403580121. doi: 10.1073/pnas.2403580121. Epub 2024 Jun 24. Proc Natl Acad Sci U S A. 2024. PMID: 38913898 Free PMC article. No abstract available.
-
DiffPaSS-high-performance differentiable pairing of protein sequences using soft scores.Bioinformatics. 2024 Dec 26;41(1):btae738. doi: 10.1093/bioinformatics/btae738. Bioinformatics. 2024. PMID: 39672677 Free PMC article.
-
Multimeric protein interaction and complex prediction: Structure, dynamics and function.Comput Struct Biotechnol J. 2025 May 16;27:1975-1997. doi: 10.1016/j.csbj.2025.05.009. eCollection 2025. Comput Struct Biotechnol J. 2025. PMID: 40496891 Free PMC article. Review.
-
Genomic language model predicts protein co-regulation and function.Nat Commun. 2024 Apr 3;15(1):2880. doi: 10.1038/s41467-024-46947-9. Nat Commun. 2024. PMID: 38570504 Free PMC article.
References
-
- Lin Z., et al. , Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
