

This is a preprint.
A nucleosome switch primes Hepatitis B Virus infection
- PMID: 38915612
- PMCID: PMC11195122
- DOI: 10.1101/2023.03.03.531011
A nucleosome switch primes Hepatitis B Virus infection
Update in
-
A nucleosome switch primes hepatitis B virus infection.Cell. 2025 Apr 17;188(8):2111-2126.e21. doi: 10.1016/j.cell.2025.01.033. Epub 2025 Feb 20. Cell. 2025. PMID: 39983728
Abstract
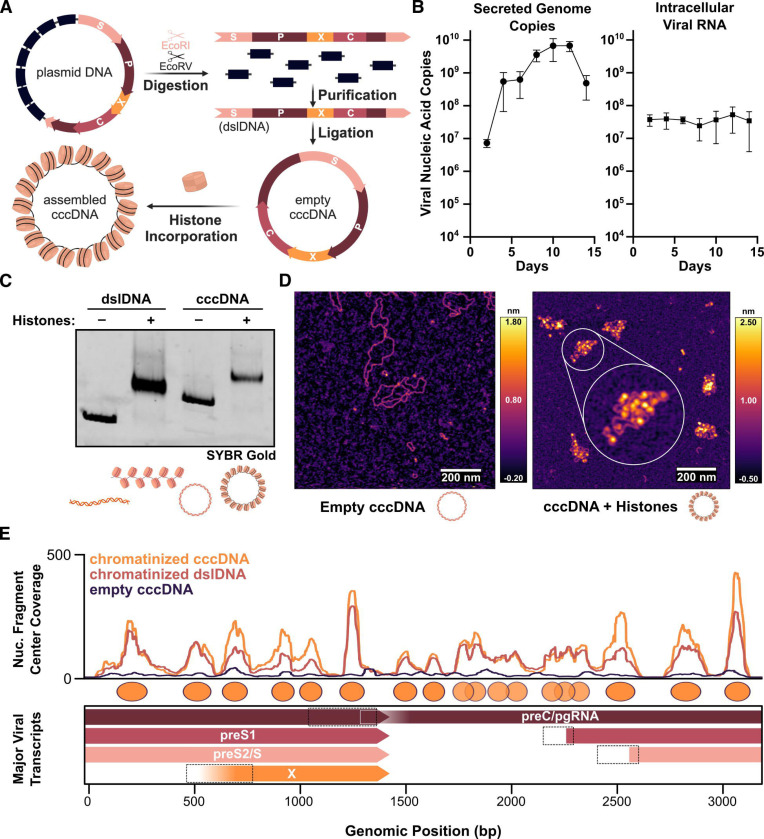
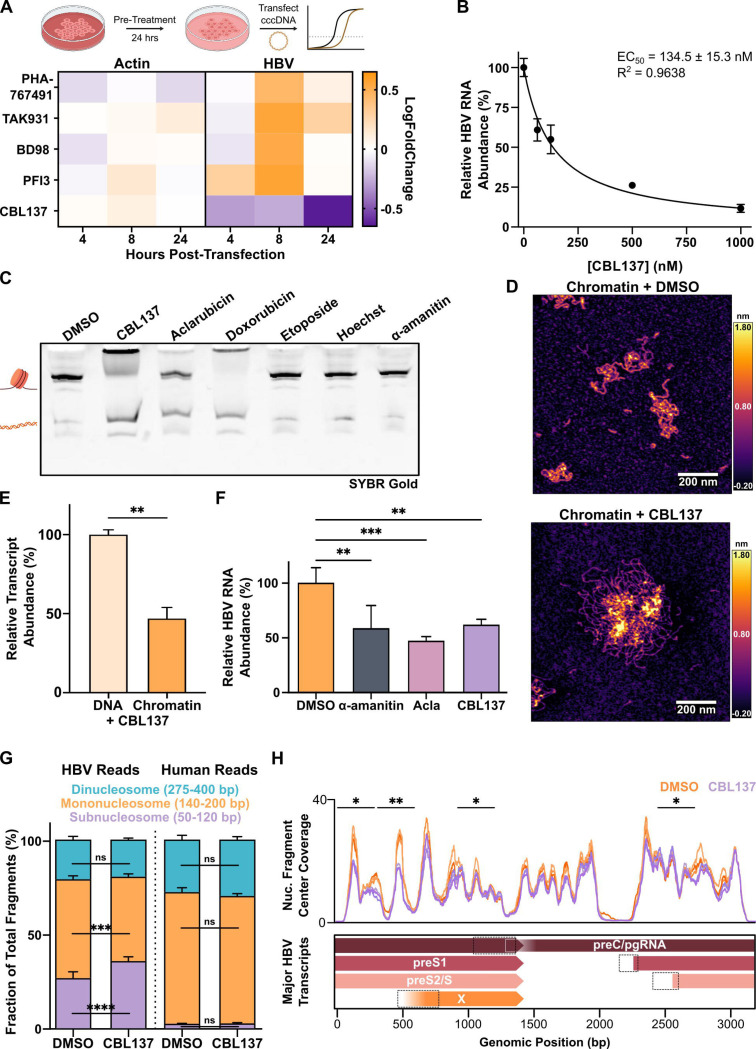
Chronic hepatitis B virus (HBV) infection is an incurable global health threat responsible for causing liver disease and hepatocellular carcinoma. During the genesis of infection, HBV establishes an independent minichromosome consisting of the viral covalently closed circular DNA (cccDNA) genome and host histones. The viral X gene must be expressed immediately upon infection to induce degradation of the host silencing factor, Smc5/6. However, the relationship between cccDNA chromatinization and X gene transcription remains poorly understood. Establishing a reconstituted viral minichromosome platform, we found that nucleosome occupancy in cccDNA drives X transcription. We corroborated these findings in cells and further showed that the chromatin destabilizing molecule CBL137 inhibits X transcription and HBV infection in hepatocytes. Our results shed light on a long-standing paradox and represent a potential new therapeutic avenue for the treatment of chronic HBV infection.
Keywords: Hepatitis B Virus; chromatin; epigenetics; nucleosome stability; transcription.
Conflict of interest statement
Declaration of interests R.E.S. is on the scientific advisory boards of Miromatrix Inc. and Lime Therapeutics and is a speaker and consultant for Alnylam Inc. All other authors declare that they have no competing interests.
Figures



References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources