

This is a preprint.
Exploring penetrance of clinically relevant variants in over 800,000 humans from the Genome Aggregation Database
- PMID: 38915639
- PMCID: PMC11195293
- DOI: 10.1101/2024.06.12.593113
Exploring penetrance of clinically relevant variants in over 800,000 humans from the Genome Aggregation Database
Update in
-
Exploring penetrance of clinically relevant variants in over 800,000 humans from the Genome Aggregation Database.Nat Commun. 2025 Oct 31;16(1):9623. doi: 10.1038/s41467-025-61698-x. Nat Commun. 2025. PMID: 41173899 Free PMC article.
Abstract
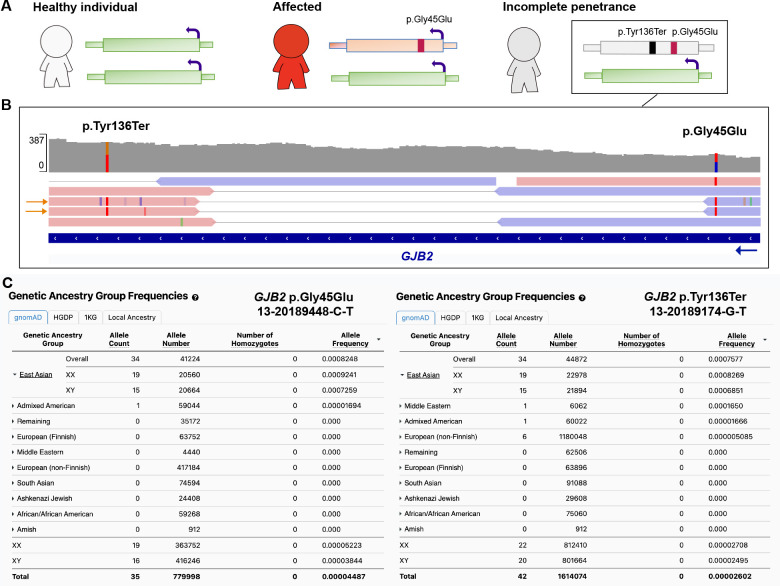
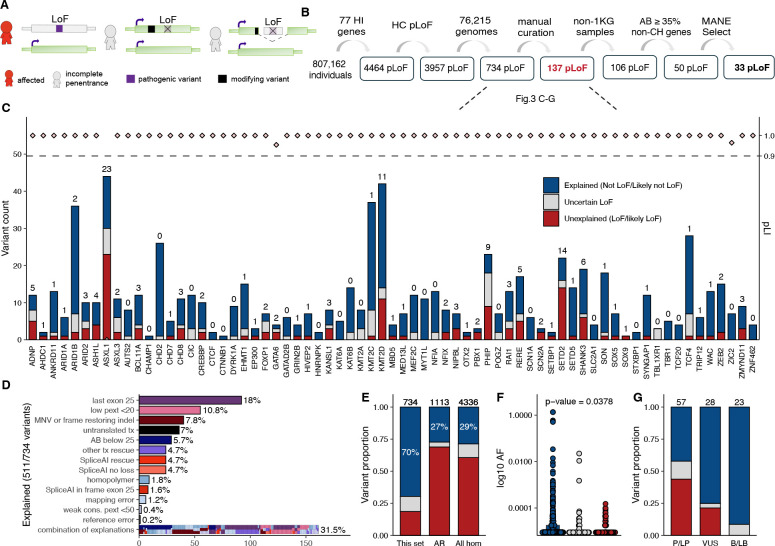
Incomplete penetrance, or absence of disease phenotype in an individual with a disease-associated variant, is a major challenge in variant interpretation. Studying individuals with apparent incomplete penetrance can shed light on underlying drivers of altered phenotype penetrance. Here, we investigate clinically relevant variants from ClinVar in 807,162 individuals from the Genome Aggregation Database (gnomAD), demonstrating improved representation in gnomAD version 4. We then conduct a comprehensive case-by-case assessment of 734 predicted loss of function variants (pLoF) in 77 genes associated with severe, early-onset, highly penetrant haploinsufficient disease. We identified explanations for the presumed lack of disease manifestation in 701 of the variants (95%). Individuals with unexplained lack of disease manifestation in this set of disorders rarely occur, underscoring the need and power of deep case-by-case assessment presented here to minimize false assignments of disease risk, particularly in unaffected individuals with higher rates of secondary properties that result in rescue.
Conflict of interest statement
COMPETING INTERESTS A.O-D.L. is on the scientific advisory board for Congenica, receives research funding in the form of reagents from Pacific Biosciences, and is a paid advisor to Addition Therapeutics and former advisor to Tome Biosciences and Ono Pharma USA. D.G.M. is a paid adviser to GlaxoSmithKline, Insitro, and Overtone Therapeutics, and receives research funding from Microsoft Corporation. H.L.R. receives research funding from Microsoft. T.L. is an advisor and has equity in Variant Bio.
Figures


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources