

OM2Seq: learning retrieval embeddings for optical genome mapping
- PMID: 38915884
- PMCID: PMC11194751
- DOI: 10.1093/bioadv/vbae079
OM2Seq: learning retrieval embeddings for optical genome mapping
Abstract
Motivation: Genomics-based diagnostic methods that are quick, precise, and economical are essential for the advancement of precision medicine, with applications spanning the diagnosis of infectious diseases, cancer, and rare diseases. One technology that holds potential in this field is optical genome mapping (OGM), which is capable of detecting structural variations, epigenomic profiling, and microbial species identification. It is based on imaging of linearized DNA molecules that are stained with fluorescent labels, that are then aligned to a reference genome. However, the computational methods currently available for OGM fall short in terms of accuracy and computational speed.
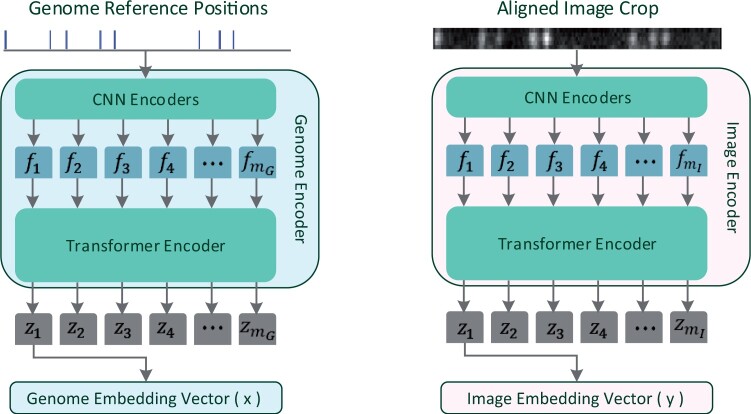
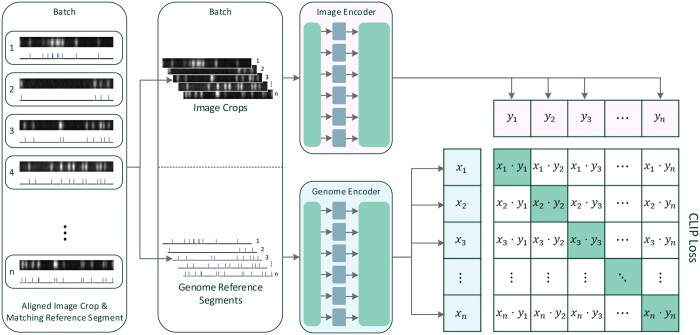
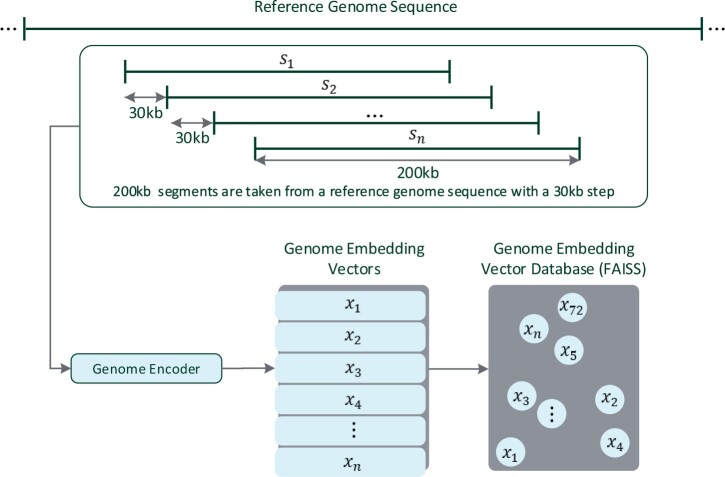
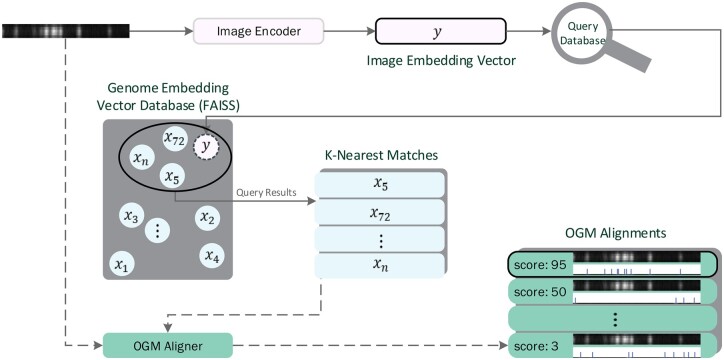
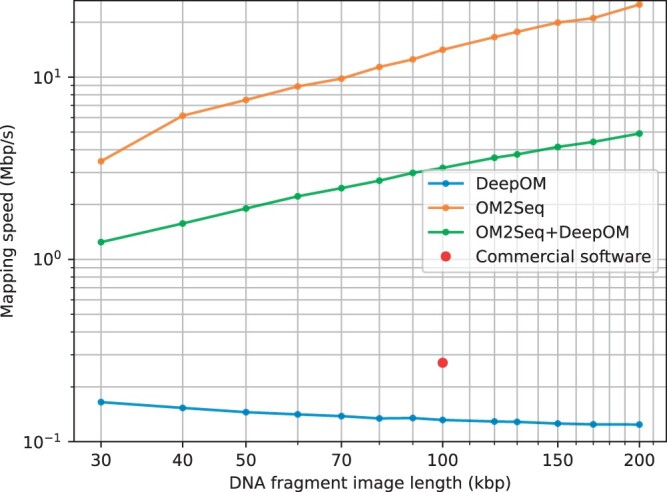
Results: This work introduces OM2Seq, a new approach for the rapid and accurate mapping of DNA fragment images to a reference genome. Based on a Transformer-encoder architecture, OM2Seq is trained on acquired OGM data to efficiently encode DNA fragment images and reference genome segments to a common embedding space, which can be indexed and efficiently queried using a vector database. We show that OM2Seq significantly outperforms the baseline methods in both computational speed (by 2 orders of magnitude) and accuracy.
Availability and implementation: https://github.com/yevgenin/om2seq.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures


References
-
- Chen S, Wang C, Chen Z. et al. Wavlm: large-scale self-supervised pre-training for full stack speech processing. IEEE J Sel Top Signal Process 2022;16:1505–18.
-
- Clopper CJ, Pearson ES.. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 1934;26:404–13.
LinkOut - more resources
Full Text Sources
