

An all-atom protein generative model
- PMID: 38916999
- PMCID: PMC11228509
- DOI: 10.1073/pnas.2311500121
An all-atom protein generative model
Abstract
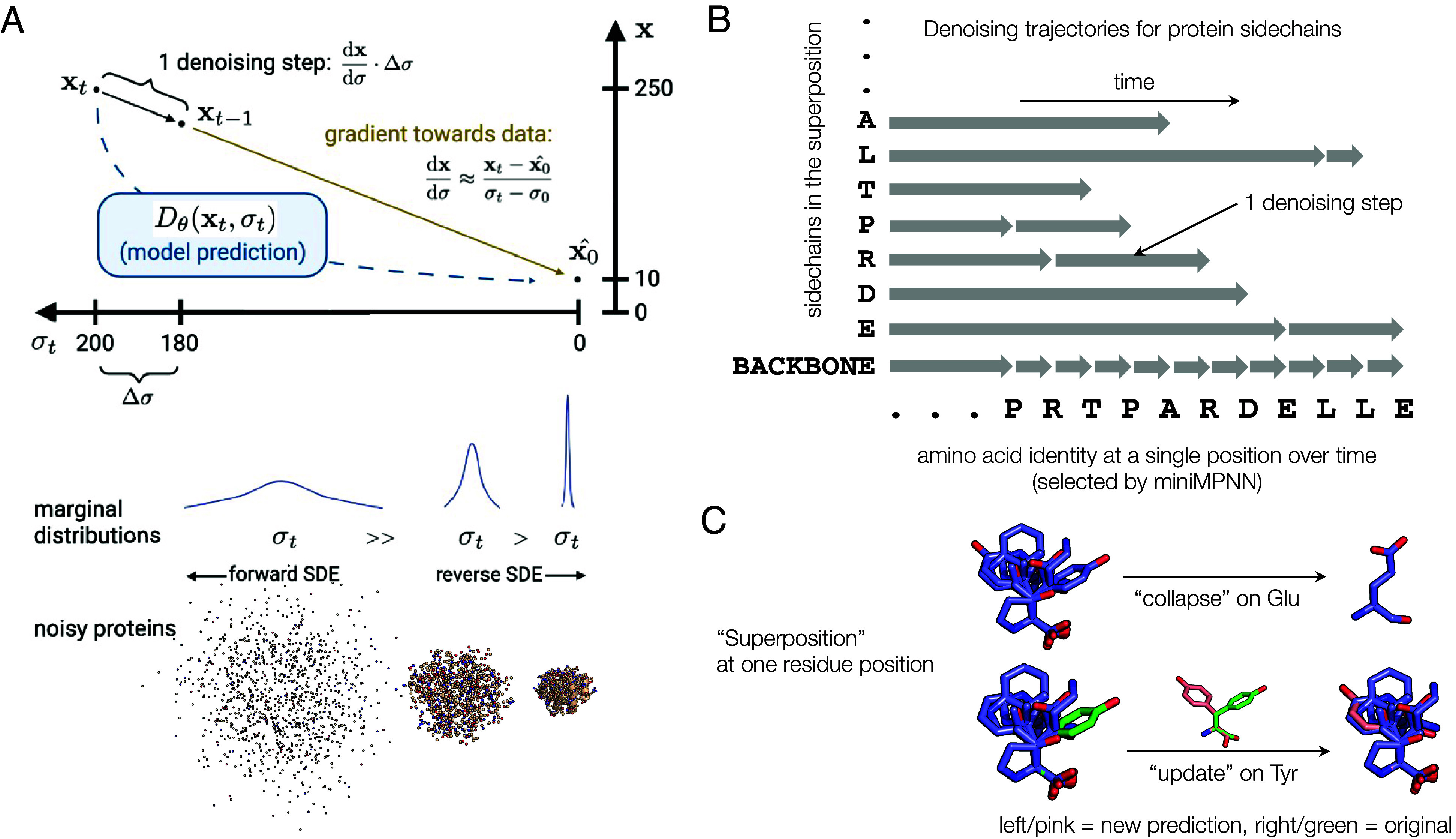
Proteins mediate their functions through chemical interactions; modeling these interactions, which are typically through sidechains, is an important need in protein design. However, constructing an all-atom generative model requires an appropriate scheme for managing the jointly continuous and discrete nature of proteins encoded in the structure and sequence. We describe an all-atom diffusion model of protein structure, Protpardelle, which represents all sidechain states at once as a "superposition" state; superpositions defining a protein are collapsed into individual residue types and conformations during sample generation. When combined with sequence design methods, our model is able to codesign all-atom protein structure and sequence. Generated proteins are of good quality under the typical quality, diversity, and novelty metrics, and sidechains reproduce the chemical features and behavior of natural proteins. Finally, we explore the potential of our model to conduct all-atom protein design and scaffold functional motifs in a backbone- and rotamer-free way.
Keywords: full-atom model; generative modeling; protein design; protein structure; sidechain generation.
Conflict of interest statement
Competing interests statement:The authors declare no competing interest.
Figures




Update of
-
An all-atom protein generative model.bioRxiv [Preprint]. 2023 May 25:2023.05.24.542194. doi: 10.1101/2023.05.24.542194. bioRxiv. 2023. Update in: Proc Natl Acad Sci U S A. 2024 Jul 2;121(27):e2311500121. doi: 10.1073/pnas.2311500121. PMID: 37292974 Free PMC article. Updated. Preprint.
References
-
- Huang P. S., Boyken S. E., Baker D., The coming of age of de novo protein design. Nature 537, 320–327 (2016). - PubMed
-
- N. Anand, P. Huang, “Generative modeling for protein structures” in Advances in Neural Information Processing Systems, S. Bengio et al., Eds. (Curran Associates, Inc., 2018), vol. 31, (2018).
-
- N. Anand, R. Eguchi, P. S. Huang, Fully differentiable full-atom protein backbone generation. ICLR 2019 Workshop DeepGenStruct (2019). https://openreview.net/forum?id=SJxnVL8YOV. Accessed 6 June 2024.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
