

Selective advantage of mutant stem cells in human clonal hematopoiesis is associated with attenuated response to inflammation and aging
- PMID: 38917807
- PMCID: PMC11512683
- DOI: 10.1016/j.stem.2024.05.010
Selective advantage of mutant stem cells in human clonal hematopoiesis is associated with attenuated response to inflammation and aging
Abstract
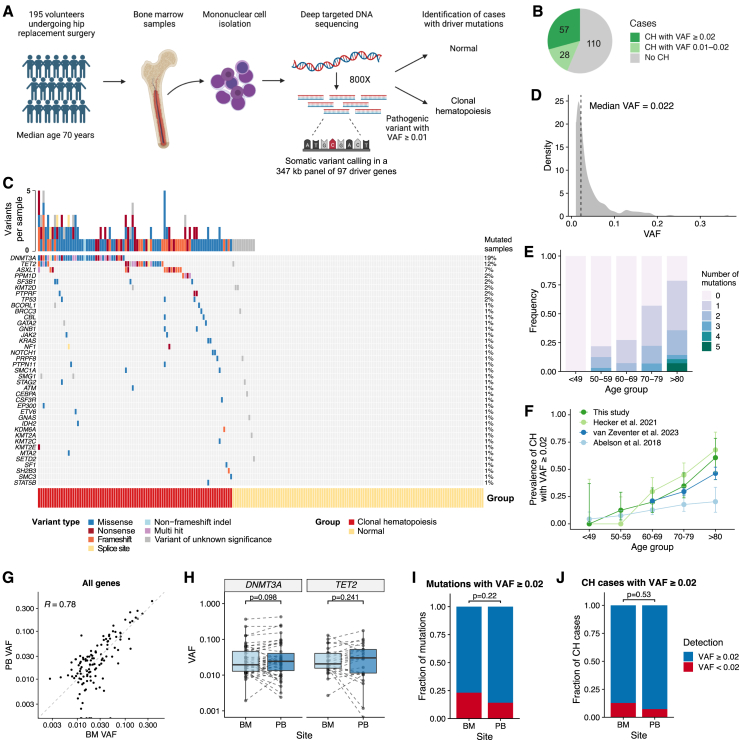
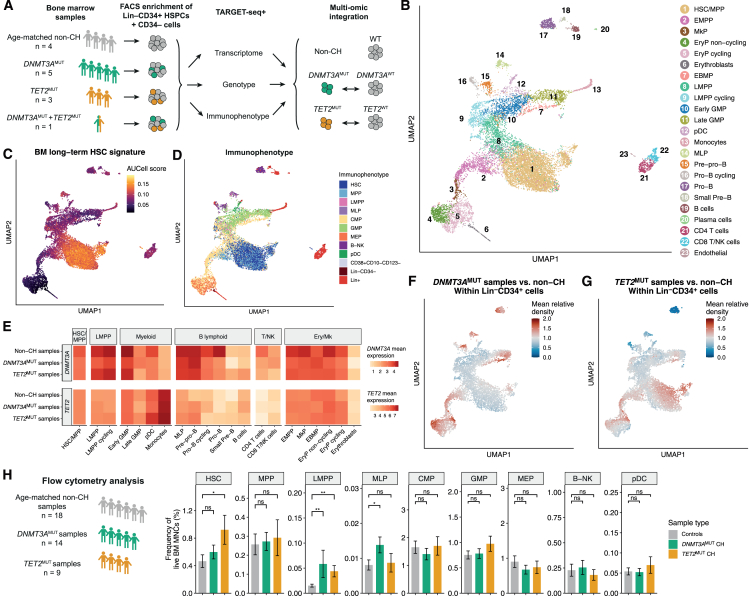
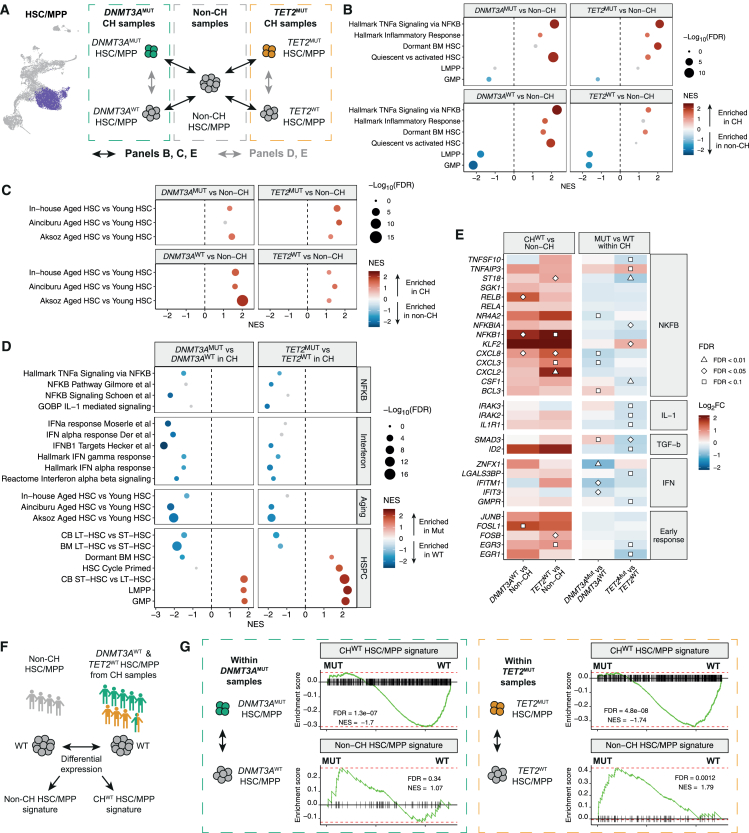
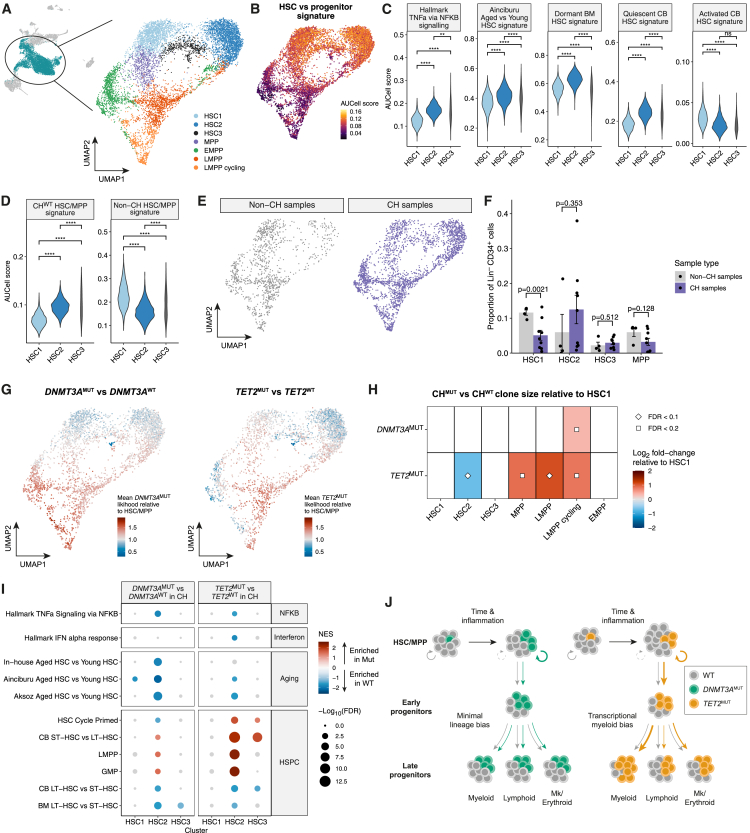
Clonal hematopoiesis (CH) arises when hematopoietic stem cells (HSCs) acquire mutations, most frequently in the DNMT3A and TET2 genes, conferring a competitive advantage through mechanisms that remain unclear. To gain insight into how CH mutations enable gradual clonal expansion, we used single-cell multi-omics with high-fidelity genotyping on human CH bone marrow (BM) samples. Most of the selective advantage of mutant cells occurs within HSCs. DNMT3A- and TET2-mutant clones expand further in early progenitors, while TET2 mutations accelerate myeloid maturation in a dose-dependent manner. Unexpectedly, both mutant and non-mutant HSCs from CH samples are enriched for inflammatory and aging transcriptomic signatures, compared with HSCs from non-CH samples, revealing a non-cell-autonomous effect. However, DNMT3A- and TET2-mutant HSCs have an attenuated inflammatory response relative to wild-type HSCs within the same sample. Our data support a model whereby CH clones are gradually selected because they are resistant to the deleterious impact of inflammation and aging.
Keywords: DNMT3A; TET2; aging; clonal competition; clonal hematopoiesis; hematopoietic stem cells; single-cell RNA-seq; single-cell genomics; somatic mosaicism.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.E.D. receives royalties from Trillium Therapeutics Inc./Pfizer and a commercial research grant from Celgene/BMS.
Figures



References
-
- Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M., et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
