

Strain-specific gut microbial signatures in type 2 diabetes identified in a cross-cohort analysis of 8,117 metagenomes
- PMID: 38918632
- PMCID: PMC11620793
- DOI: 10.1038/s41591-024-03067-7
Strain-specific gut microbial signatures in type 2 diabetes identified in a cross-cohort analysis of 8,117 metagenomes
Abstract
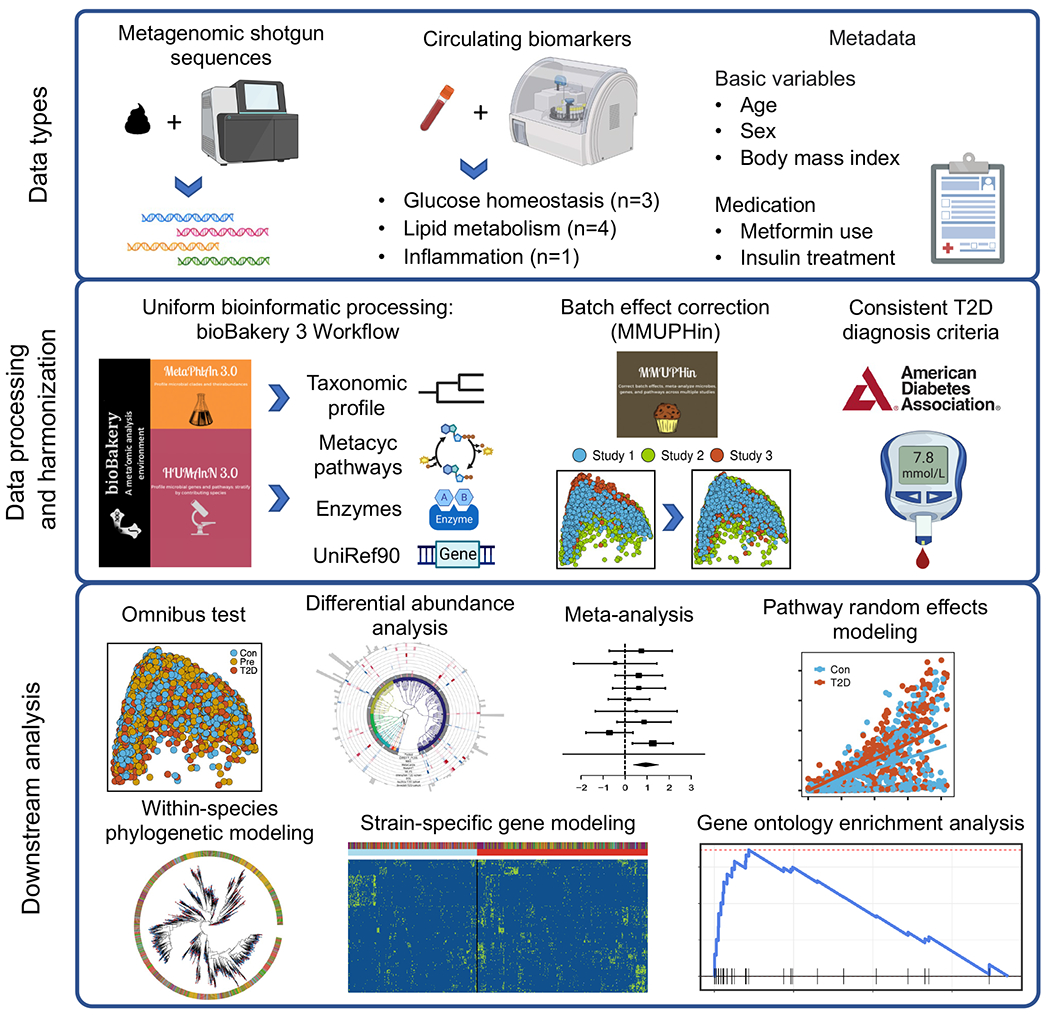
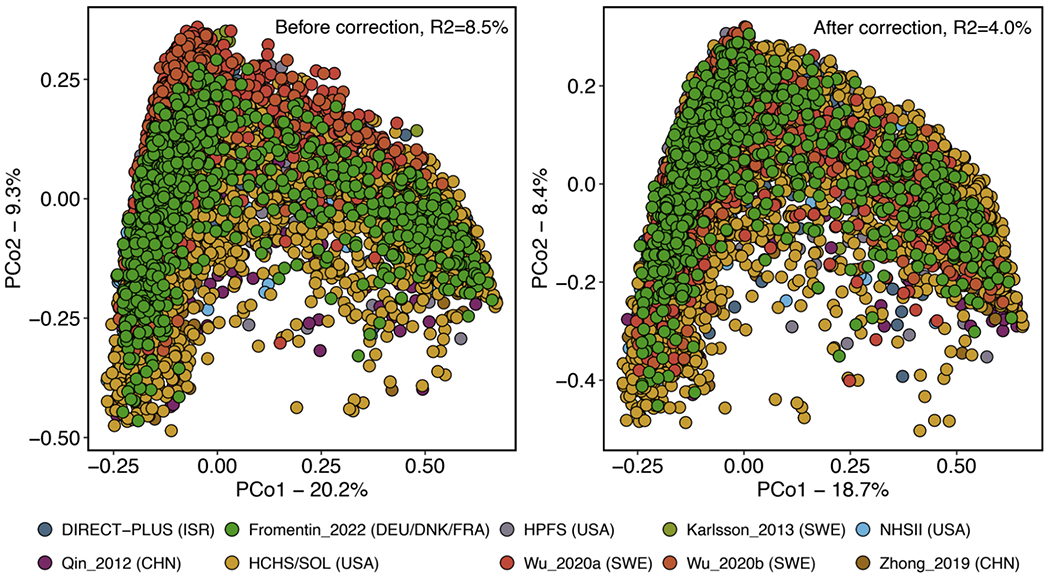
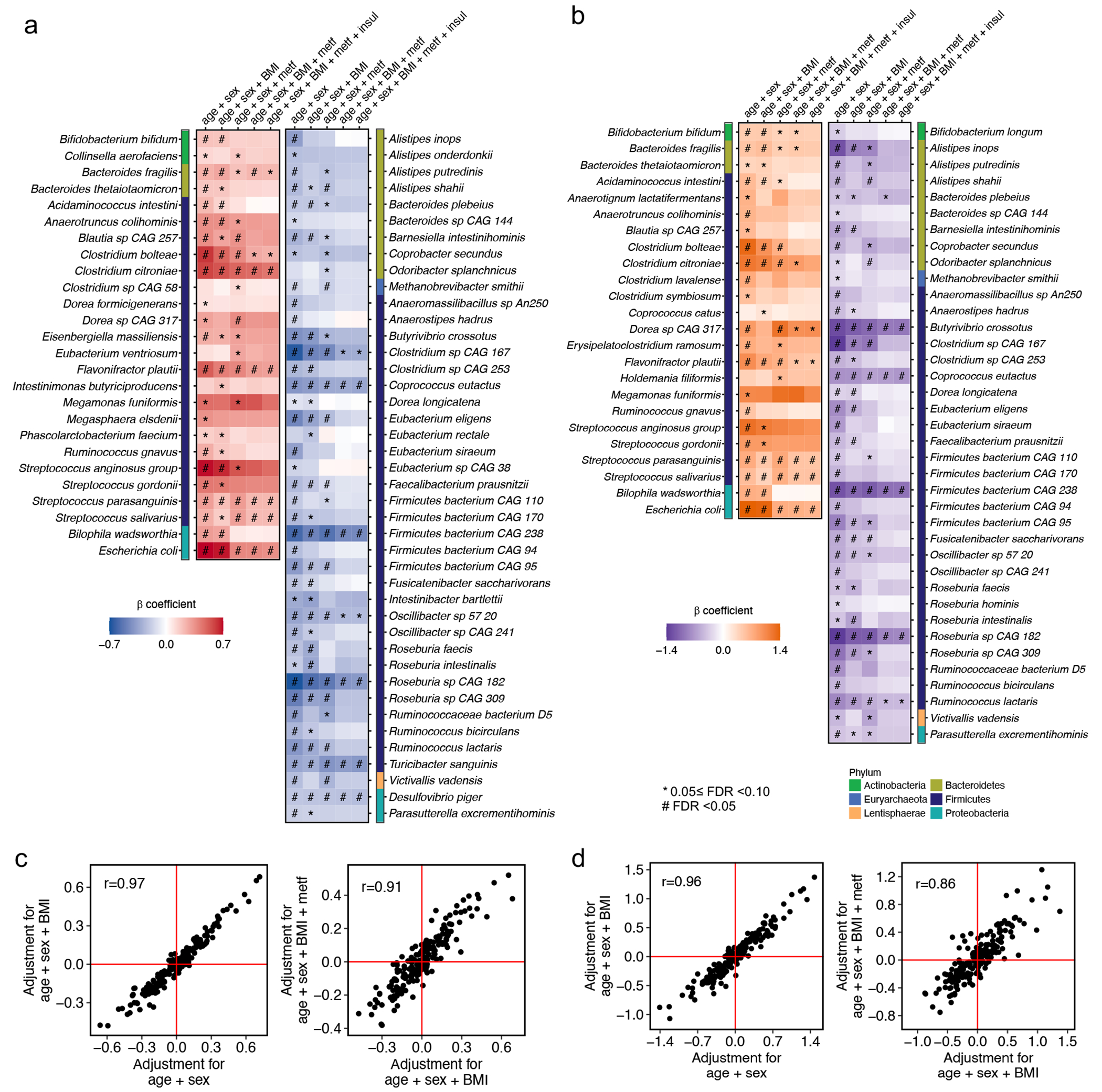
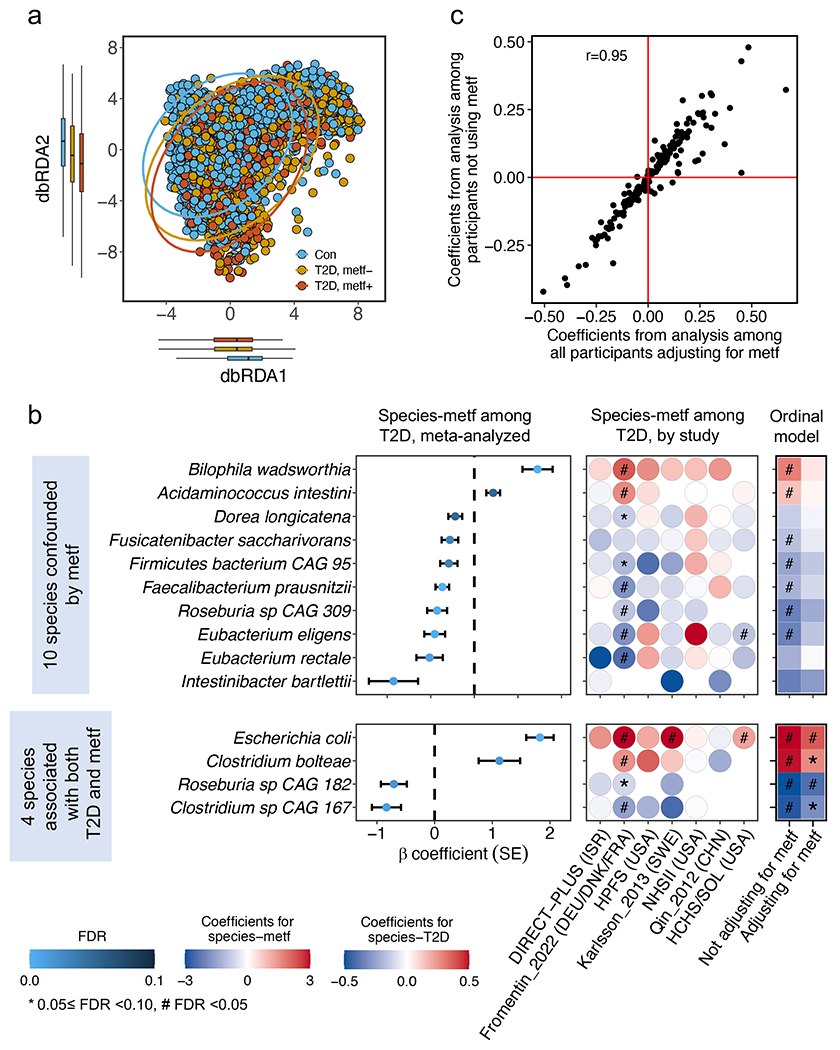
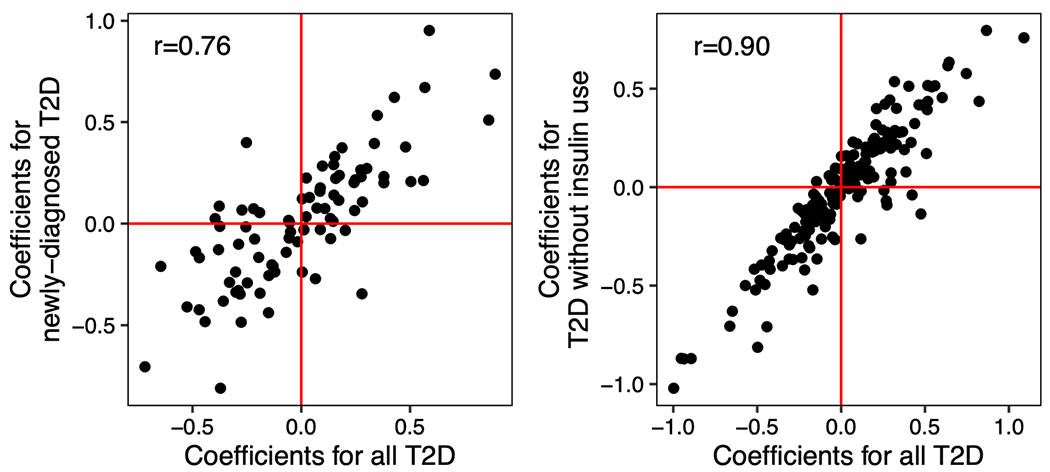
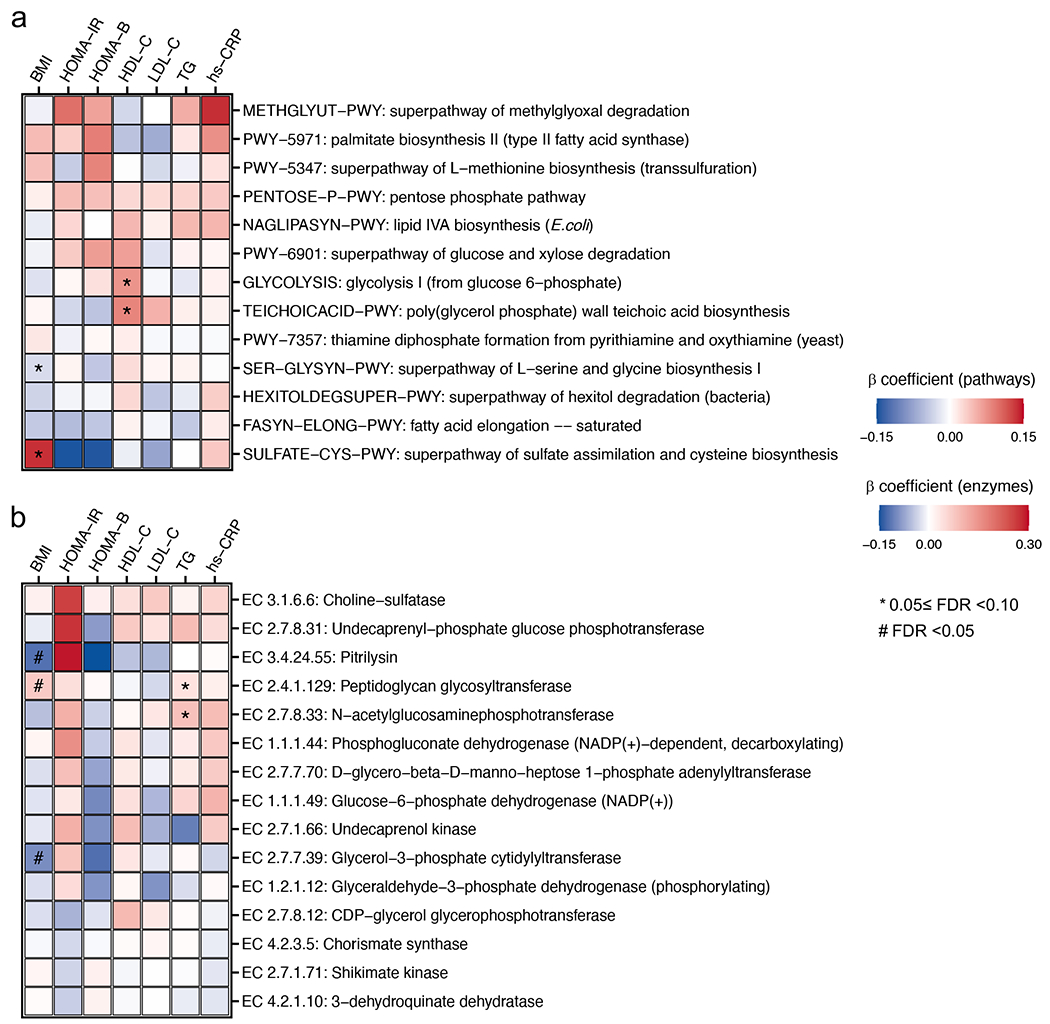
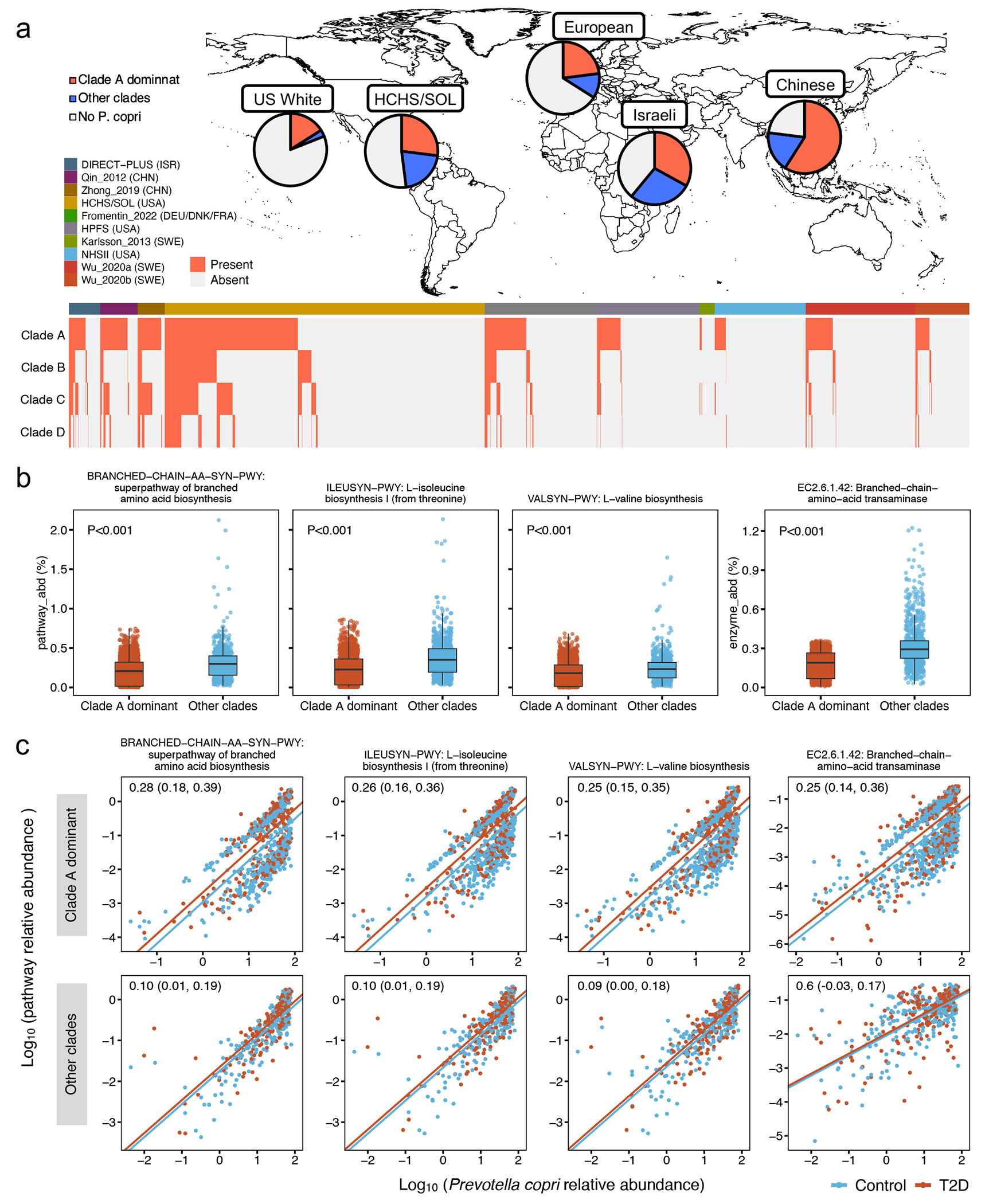
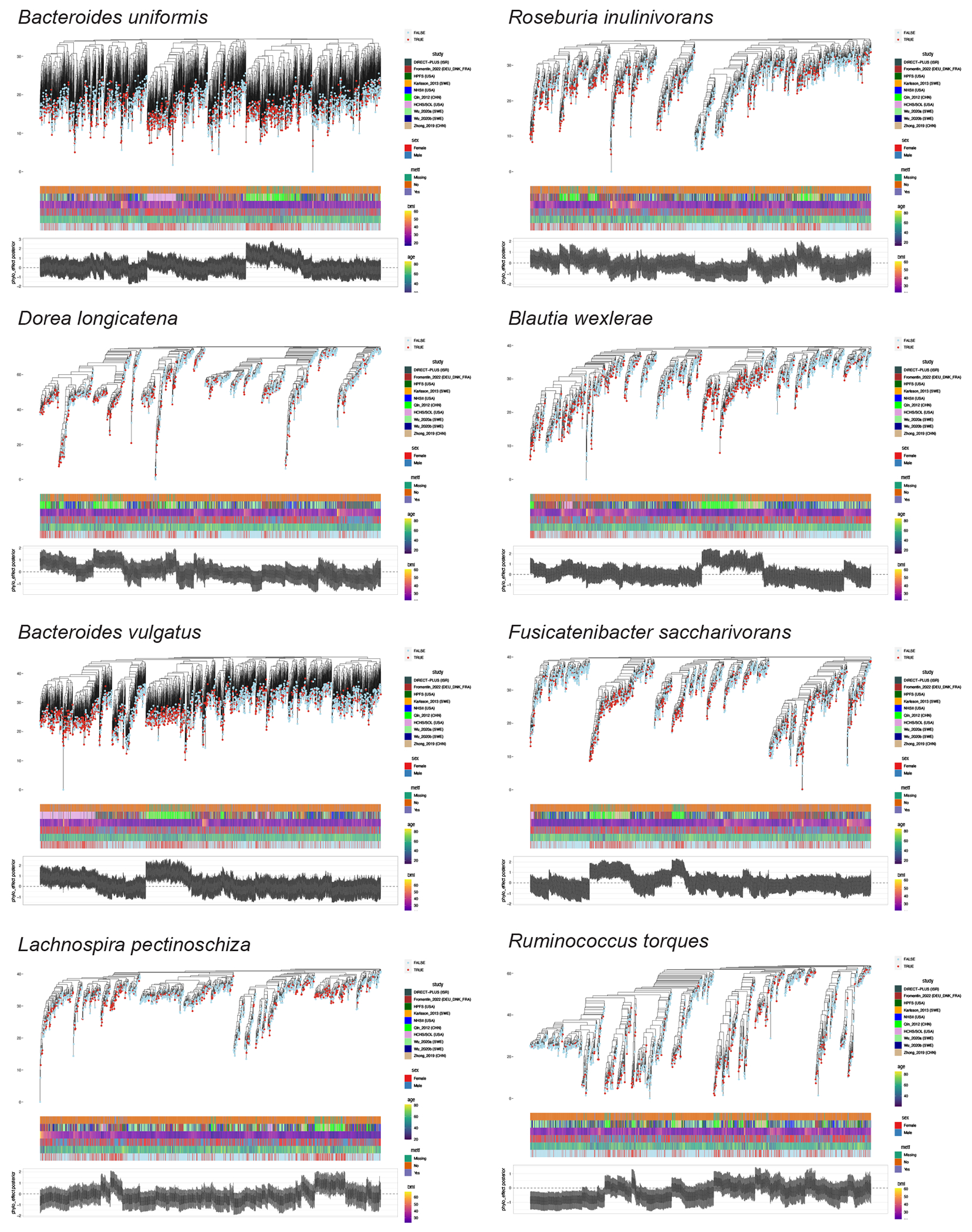
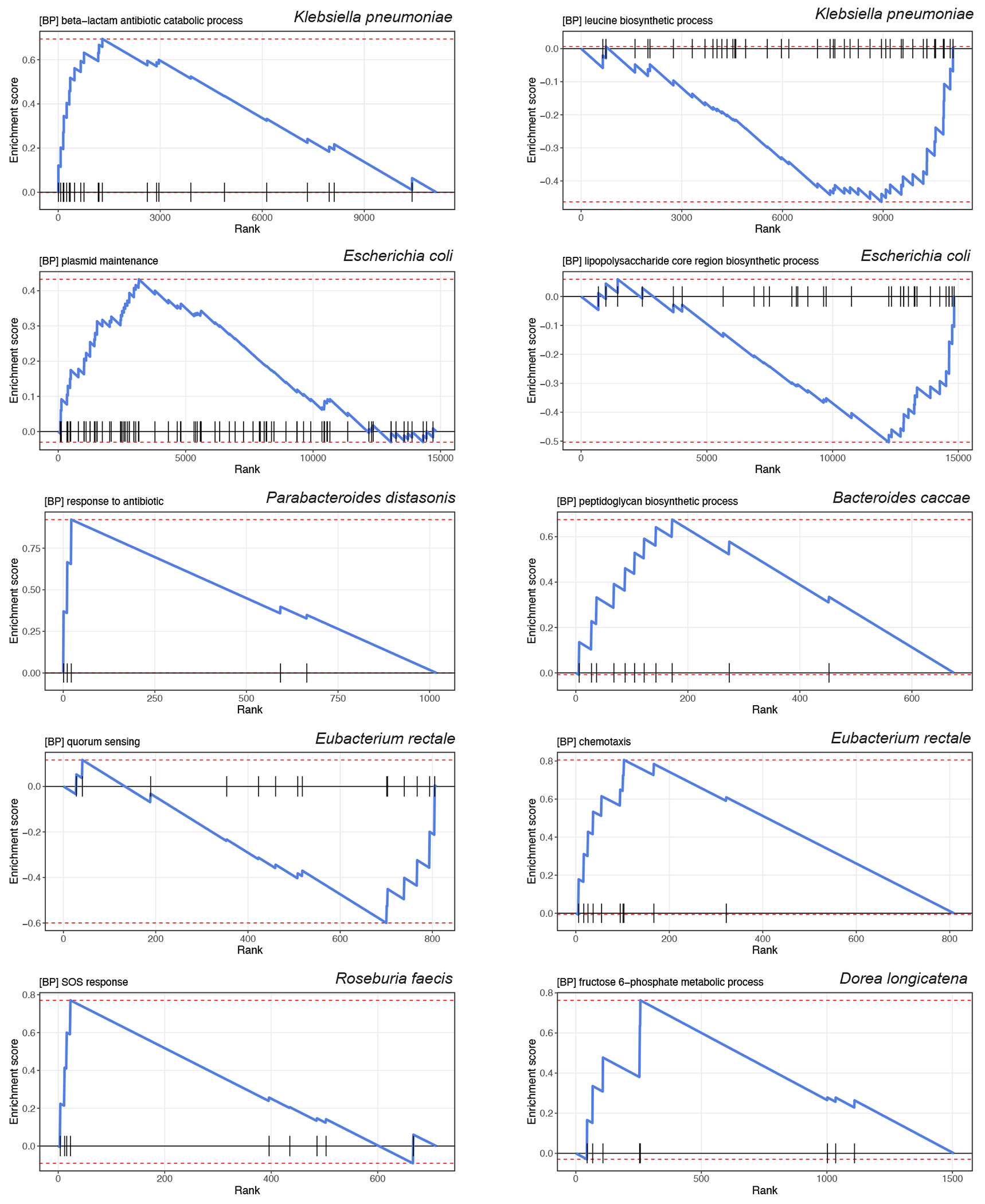
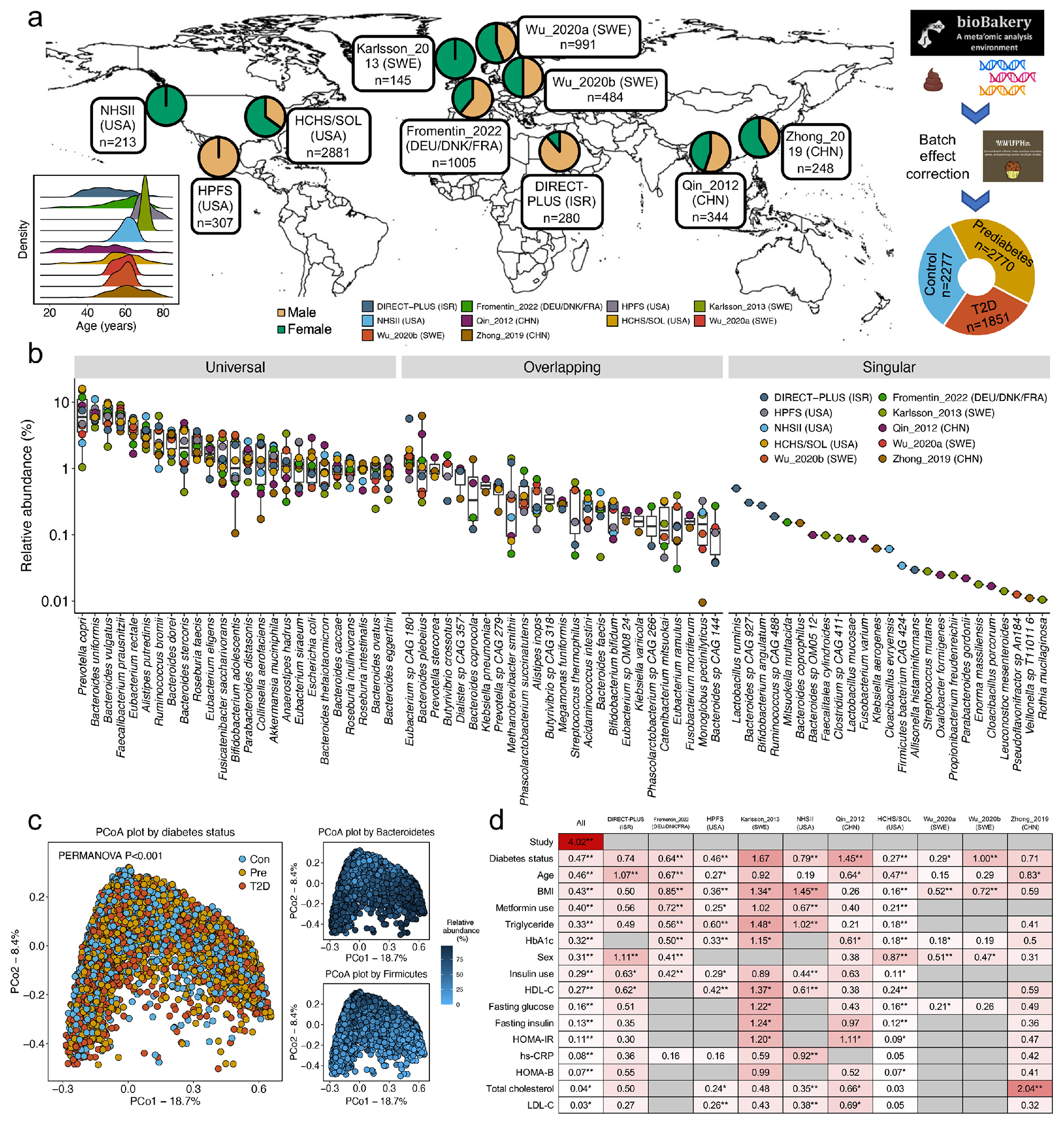
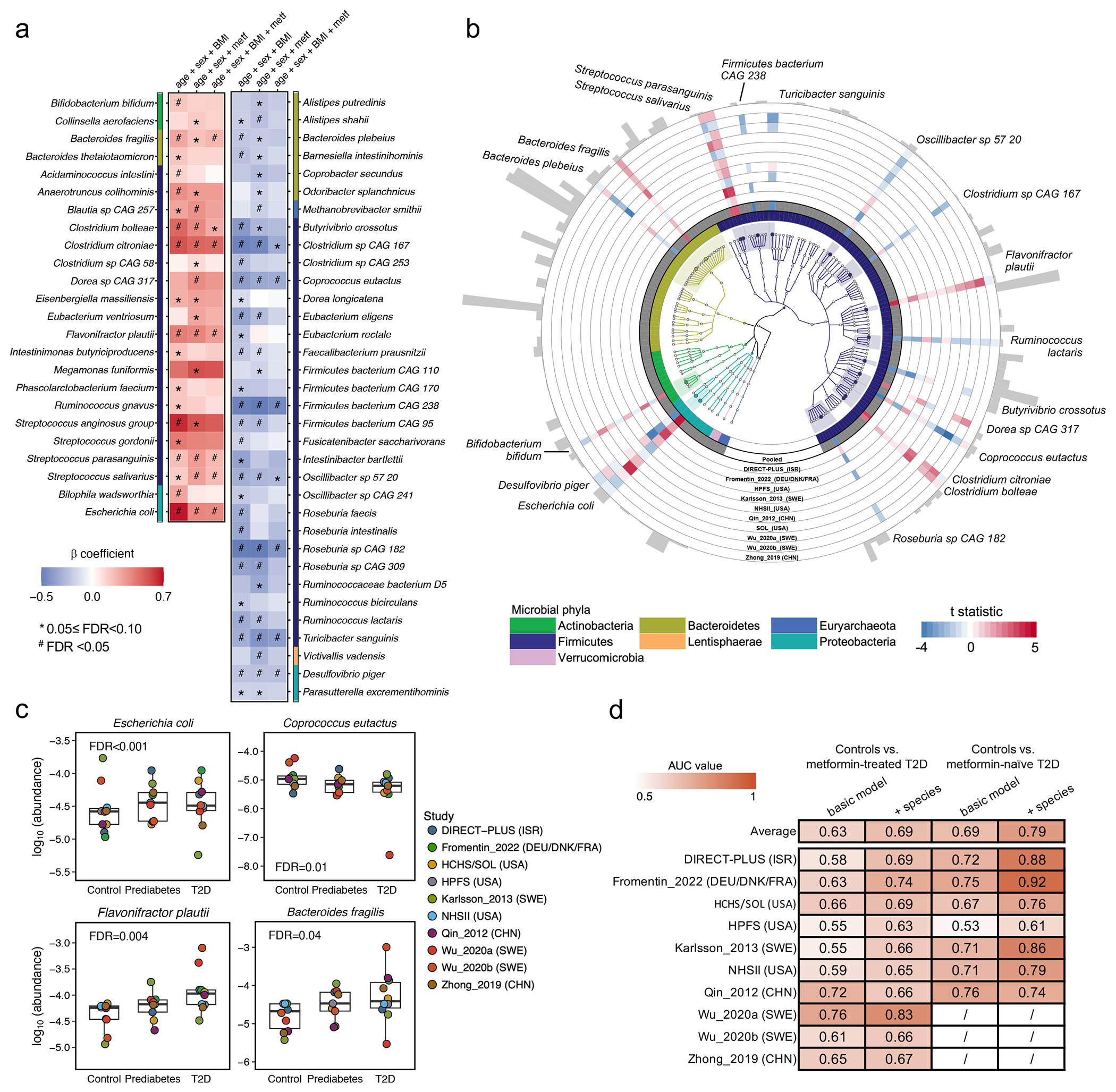
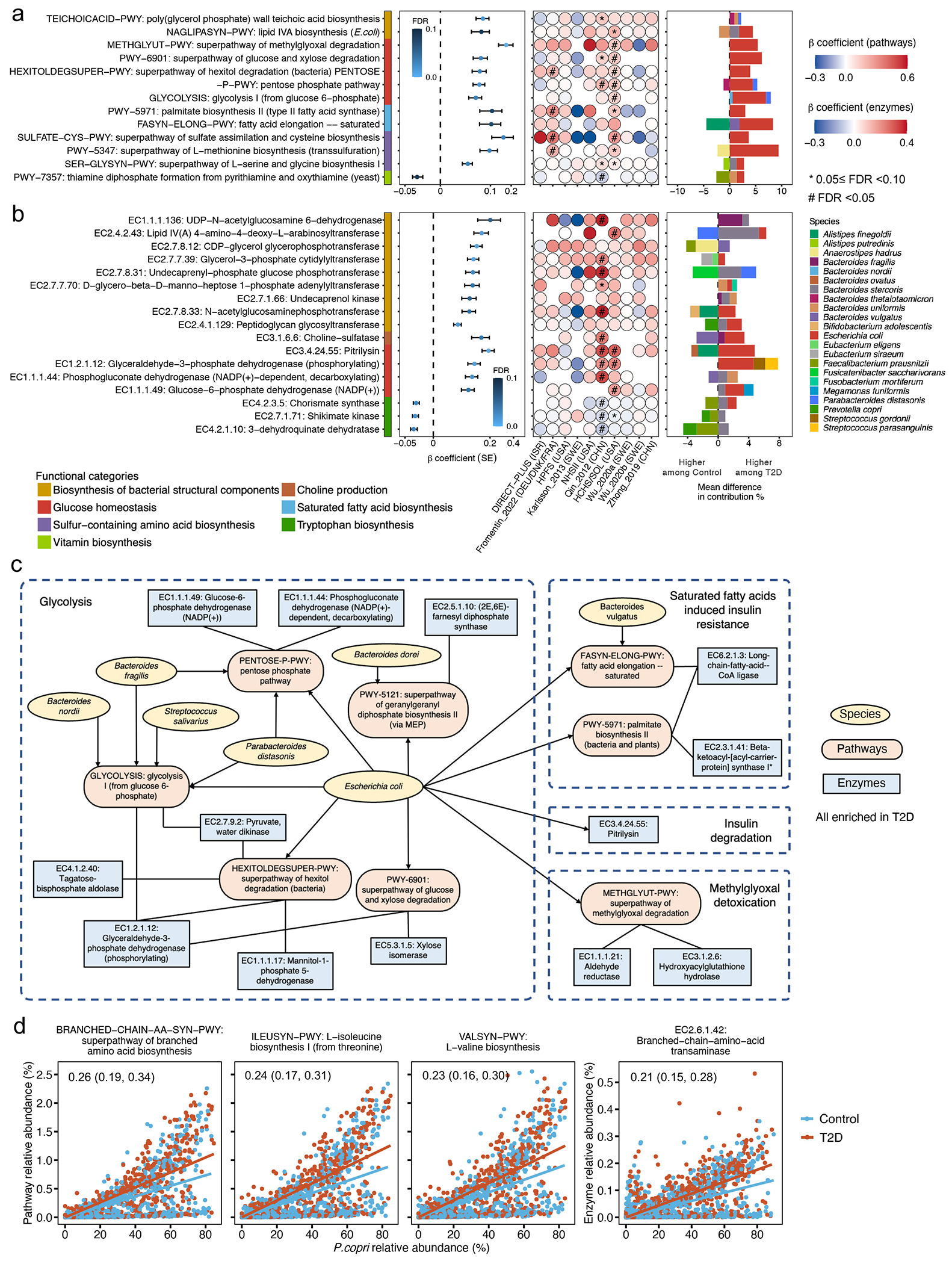
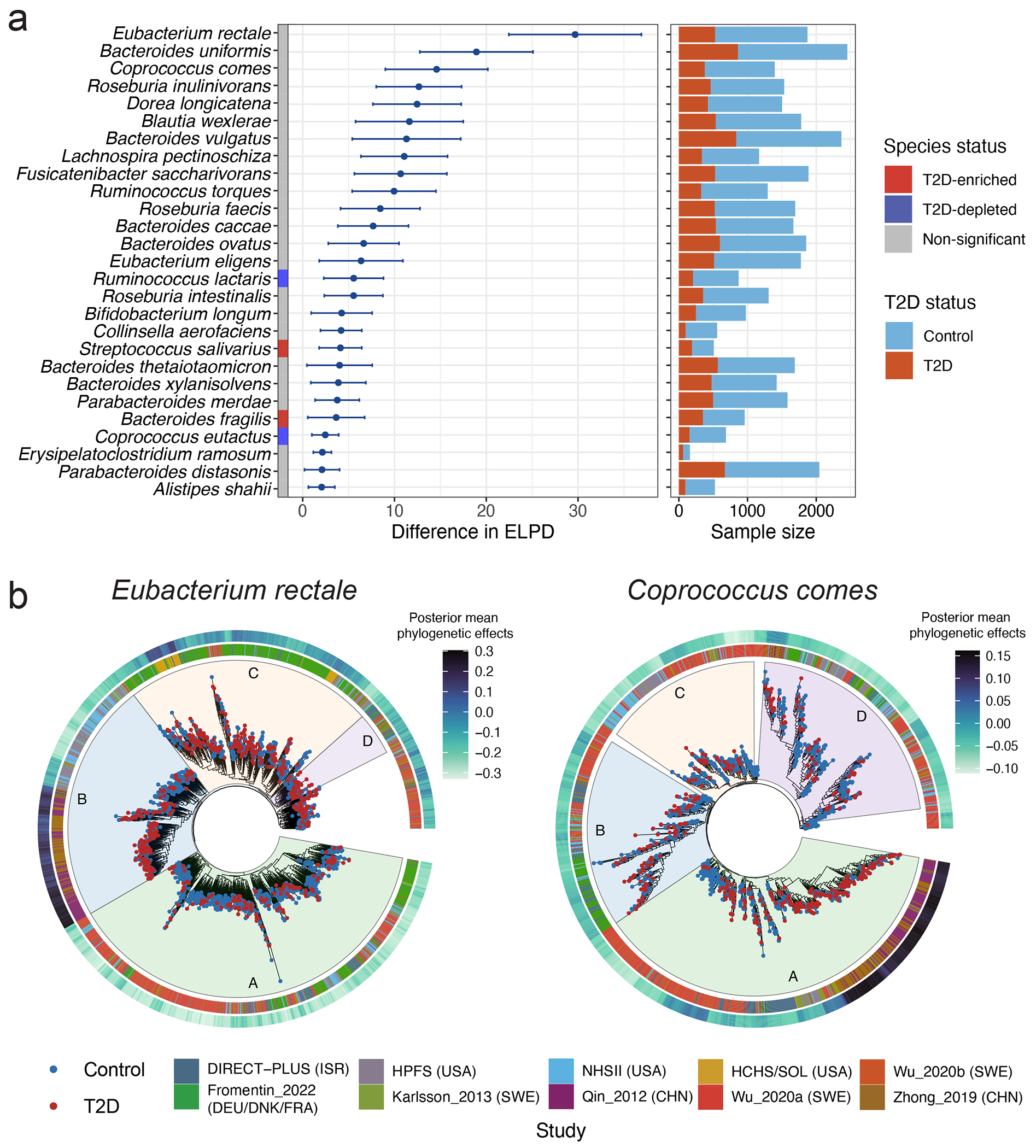
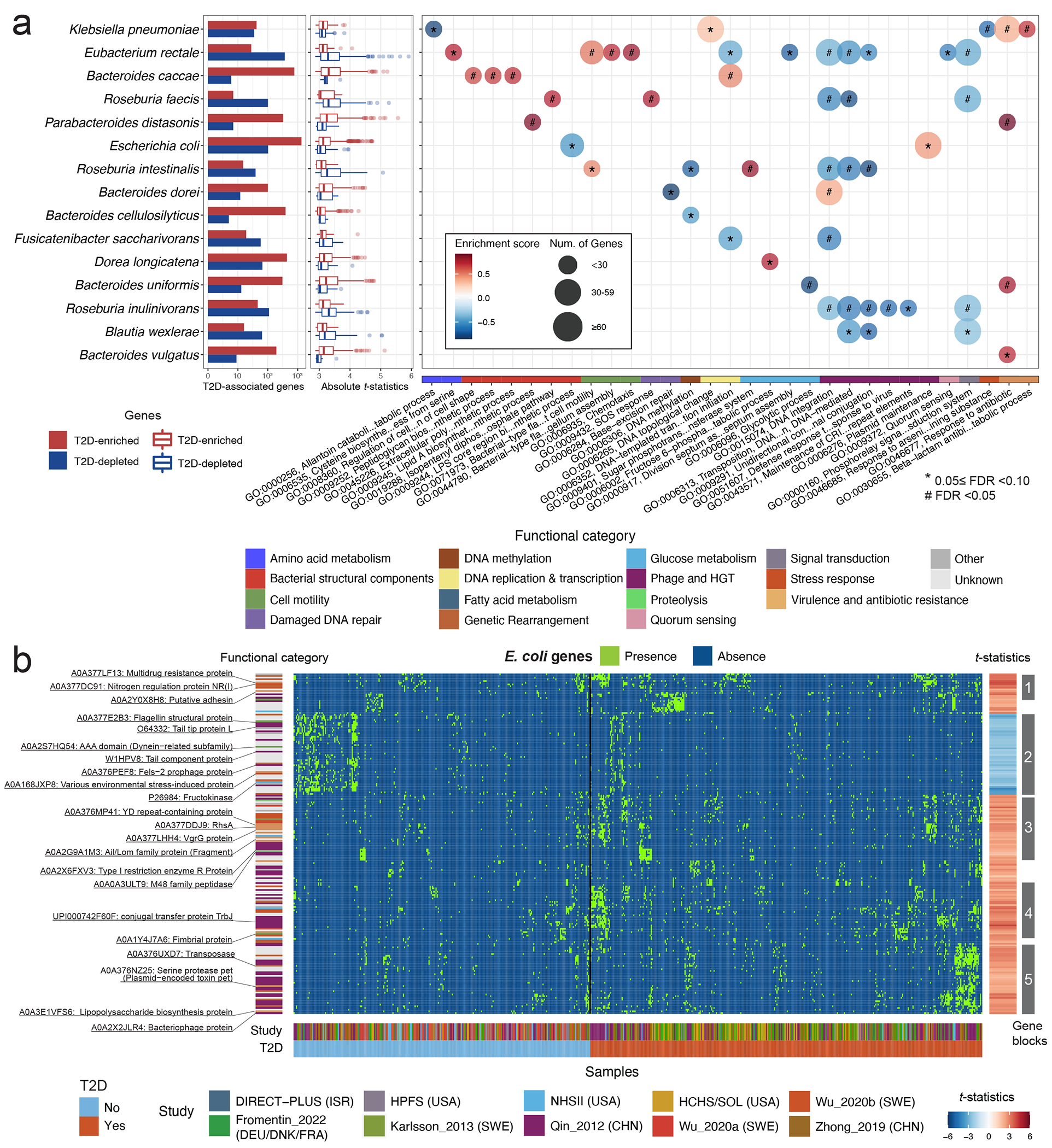
The association of gut microbial features with type 2 diabetes (T2D) has been inconsistent due in part to the complexity of this disease and variation in study design. Even in cases in which individual microbial species have been associated with T2D, mechanisms have been unable to be attributed to these associations based on specific microbial strains. We conducted a comprehensive study of the T2D microbiome, analyzing 8,117 shotgun metagenomes from 10 cohorts of individuals with T2D, prediabetes, and normoglycemic status in the United States, Europe, Israel and China. Dysbiosis in 19 phylogenetically diverse species was associated with T2D (false discovery rate < 0.10), for example, enriched Clostridium bolteae and depleted Butyrivibrio crossotus. These microorganisms also contributed to community-level functional changes potentially underlying T2D pathogenesis, for example, perturbations in glucose metabolism. Our study identifies within-species phylogenetic diversity for strains of 27 species that explain inter-individual differences in T2D risk, such as Eubacterium rectale. In some cases, these were explained by strain-specific gene carriage, including loci involved in various mechanisms of horizontal gene transfer and novel biological processes underlying metabolic risk, for example, quorum sensing. In summary, our study provides robust cross-cohort microbial signatures in a strain-resolved manner and offers new mechanistic insights into T2D.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
C.H. is a member of the scientific advisory board for Zoe Nutrition, Empress Therapeutics, and Seres Therapeutics. The other authors declare no competing interests.
Figures
References
-
- IDF Diabetes Atlas 2021. (https://diabetesatlas.org/atlas/tenth-edition/).
-
- American Diabetes Association Professional Practice, C. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care 45, S17–S38 (2022). - PubMed
-
- Canfora EE, Meex RCR, Venema K & Blaak EE Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol 15, 261–273 (2019). - PubMed
-
- Karlsson FH, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103 (2013). - PubMed
MeSH terms
Grants and funding
- K99 DK119412/DK/NIDDK NIH HHS/United States
- R01NR01999/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U01 CA152904/CA/NCI NIH HHS/United States
- U01 CA167552/CA/NCI NIH HHS/United States
- R01AG077489/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 NR019992/NR/NINR NIH HHS/United States
- P30 DK046200/DK/NIDDK NIH HHS/United States
- R01 AG077489/AG/NIA NIH HHS/United States
- RF1 AG083764/AG/NIA NIH HHS/United States
- R01 HL035464/HL/NHLBI NIH HHS/United States
- P30DK046200/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R00DK119412/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R00 DK119412/DK/NIDDK NIH HHS/United States
- N01 HC065233/HL/NHLBI NIH HHS/United States
- R01 MD011389/MD/NIMHD NIH HHS/United States
- R24DK110499/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
- N01 HC065236/HL/NHLBI NIH HHS/United States
- R01 CA202704/CA/NCI NIH HHS/United States
- P01 CA055075/CA/NCI NIH HHS/United States
- N01 HC065235/HL/NHLBI NIH HHS/United States
- 209933838/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 897161/American Heart Association (American Heart Association, Inc.)
- U01 CA176726/CA/NCI NIH HHS/United States
- P30 DK111022/DK/NIDDK NIH HHS/United States
- N01 HC065234/HL/NHLBI NIH HHS/United States
- R35 CA253185/CA/NCI NIH HHS/United States
- R01 HL060712/HL/NHLBI NIH HHS/United States
- N01 HC065237/HL/NHLBI NIH HHS/United States
- R24 DK110499/DK/NIDDK NIH HHS/United States
