

The N6-methyladenosine Epitranscriptomic Landscape of Lung Adenocarcinoma
- PMID: 38922581
- PMCID: PMC11528209
- DOI: 10.1158/2159-8290.CD-23-1212
The N6-methyladenosine Epitranscriptomic Landscape of Lung Adenocarcinoma
Abstract
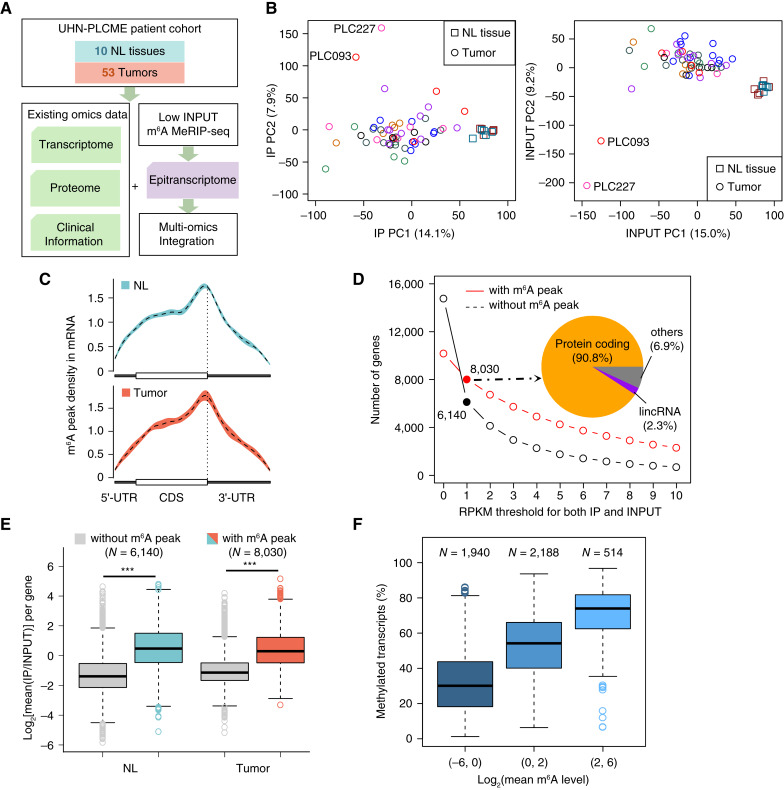
Comprehensive N6-methyladenosine (m6A) epitranscriptomic profiling of primary tumors remains largely uncharted. Here, we profiled the m6A epitranscriptome of 10 nonneoplastic lung tissues and 51 lung adenocarcinoma (LUAD) tumors, integrating the corresponding transcriptomic, proteomic, and extensive clinical annotations. We identified distinct clusters and genes that were exclusively linked to disease progression through m6A modifications. In comparison with nonneoplastic lung tissues, we identified 430 transcripts to be hypo-methylated and 222 to be hyper-methylated in tumors. Among these genes, EML4 emerged as a novel metastatic driver, displaying significant hypermethylation in tumors. m6A modification promoted the translation of EML4, leading to its widespread overexpression in primary tumors. Functionally, EML4 modulated cytoskeleton dynamics by interacting with ARPC1A, enhancing lamellipodia formation, cellular motility, local invasion, and metastasis. Clinically, high EML4 protein abundance correlated with features of metastasis. METTL3 small-molecule inhibitor markedly diminished both EML4 m6A and protein abundance and efficiently suppressed lung metastases in vivo. Significance: Our study reveals a dynamic and functional epitranscriptomic landscape in LUAD, offering a valuable resource for further research in the field. We identified EML4 hypermethylation as a key driver of tumor metastasis, highlighting a novel therapeutic strategy of targeting EML4 to prevent LUAD metastasis.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
No disclosures were reported.
Figures






References
-
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
