

MRE11 and TREX1 control senescence by coordinating replication stress and interferon signaling
- PMID: 38926338
- PMCID: PMC11208572
- DOI: 10.1038/s41467-024-49740-w
MRE11 and TREX1 control senescence by coordinating replication stress and interferon signaling
Abstract
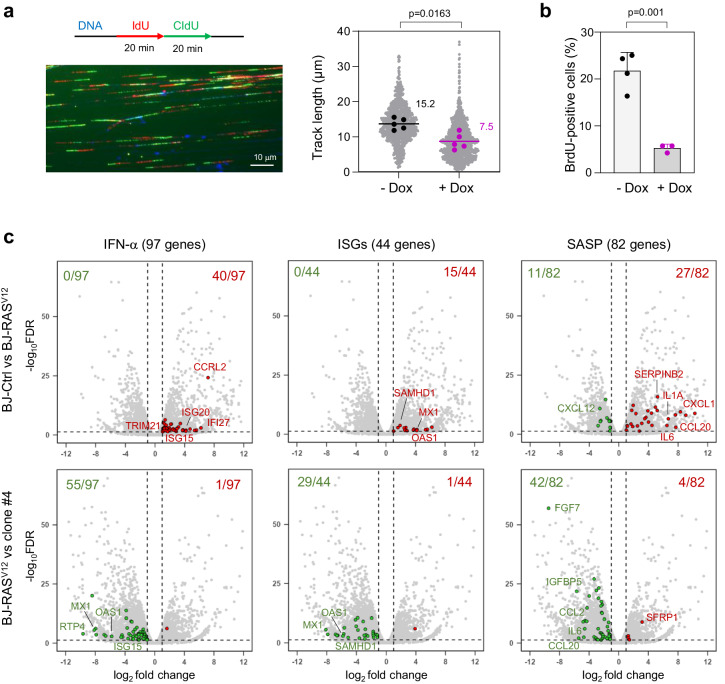
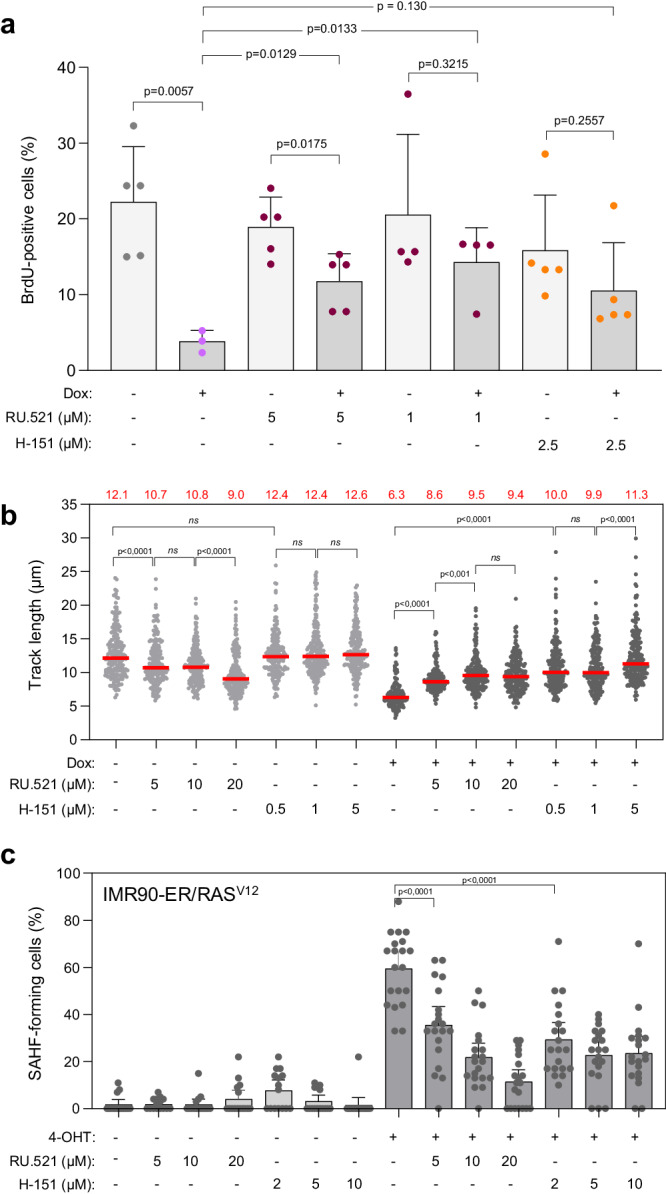
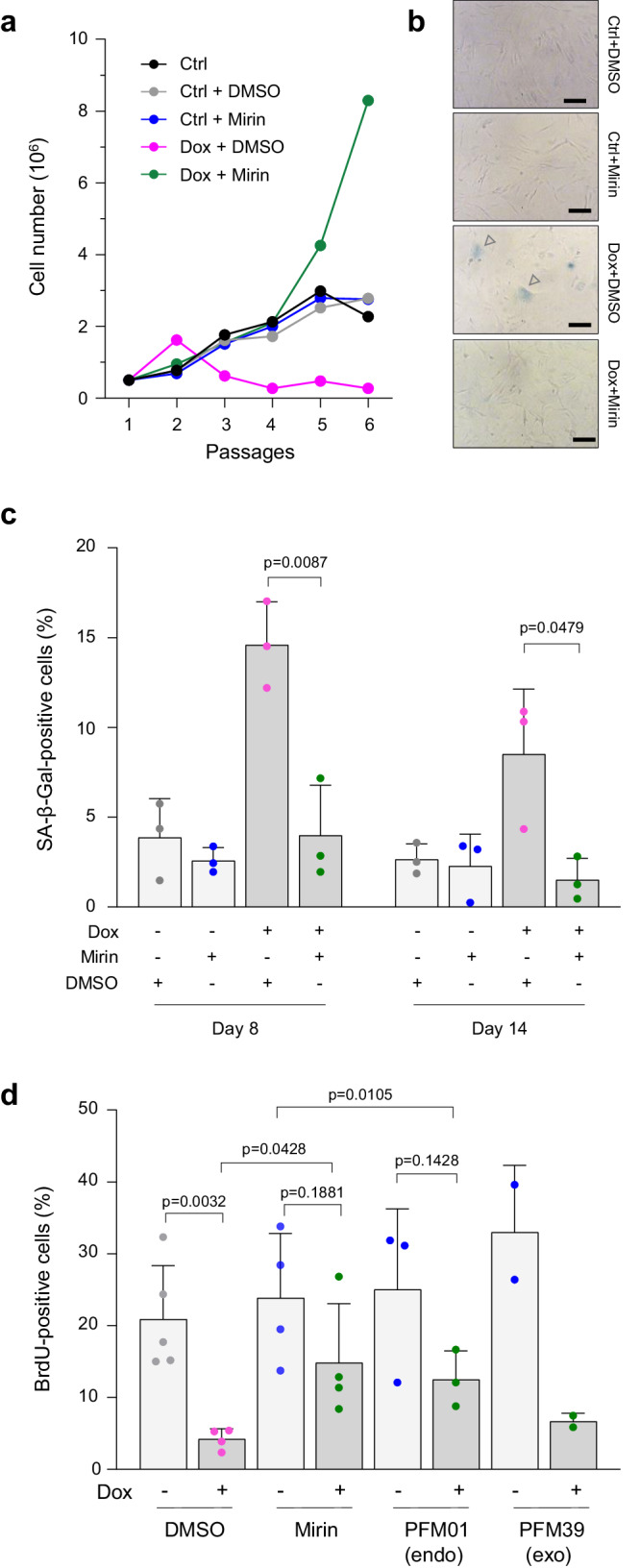
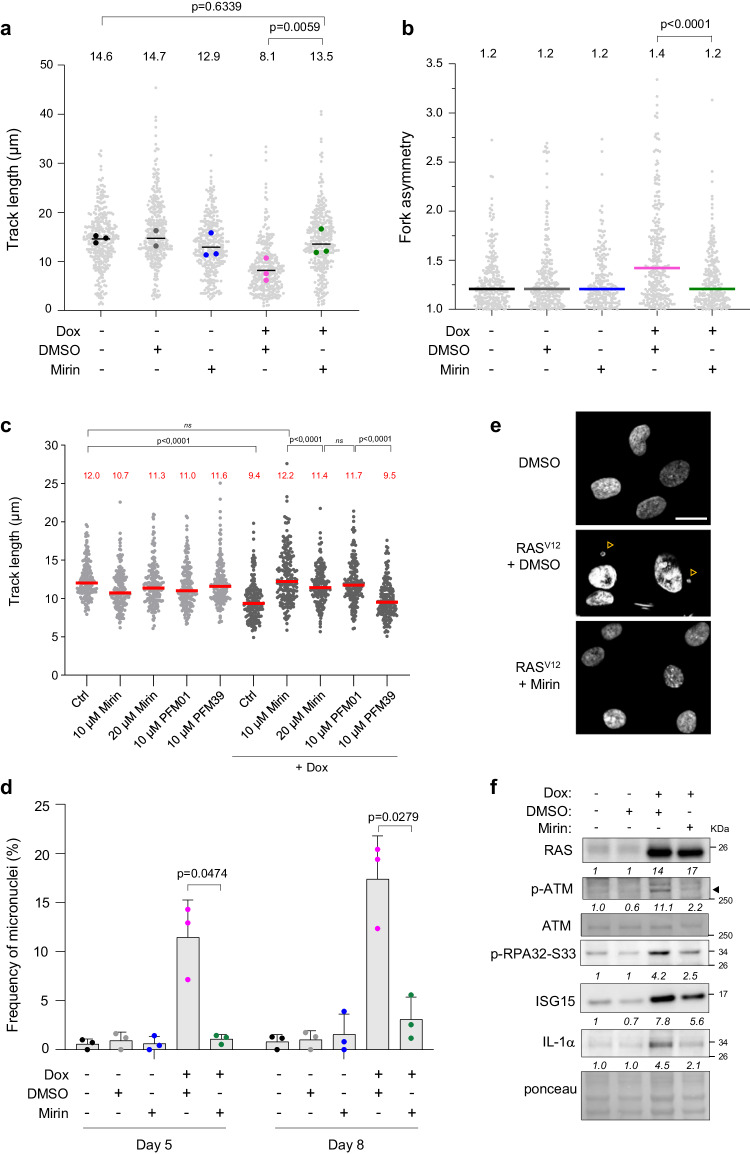
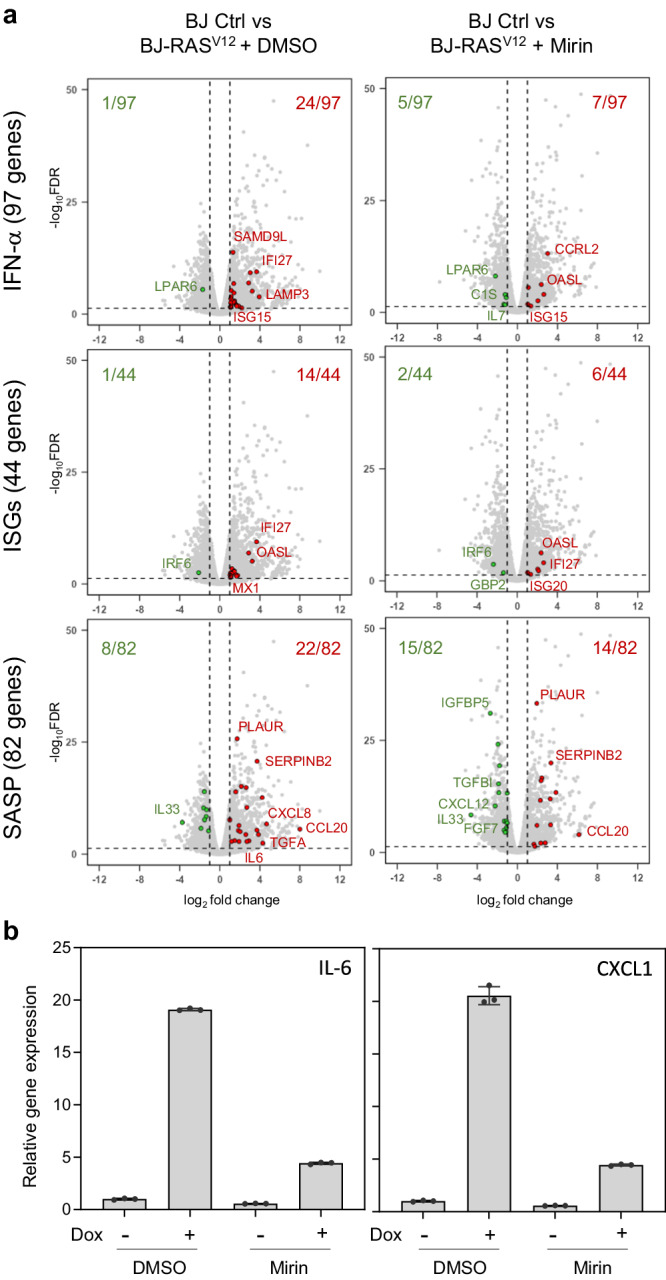
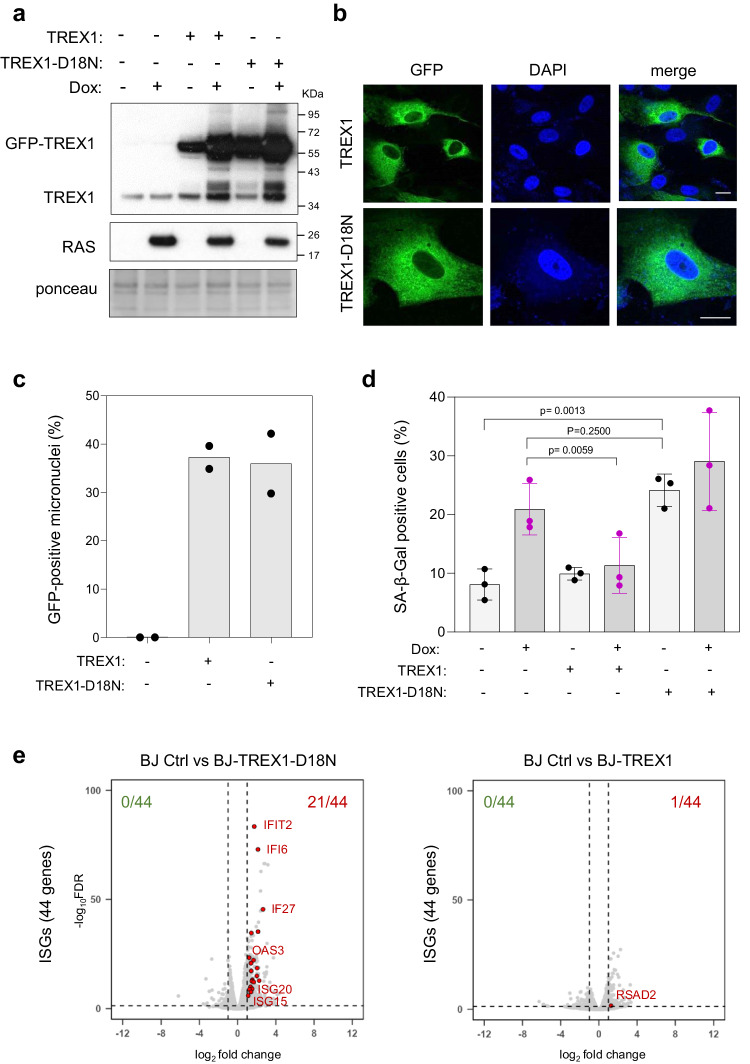
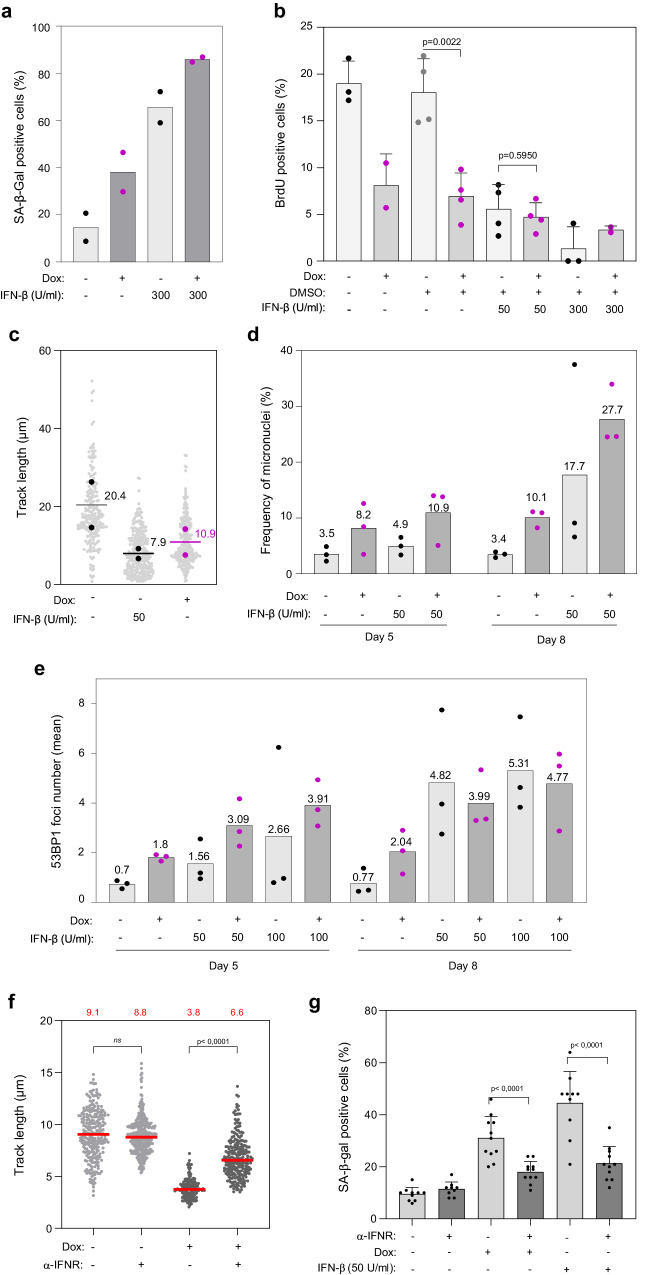
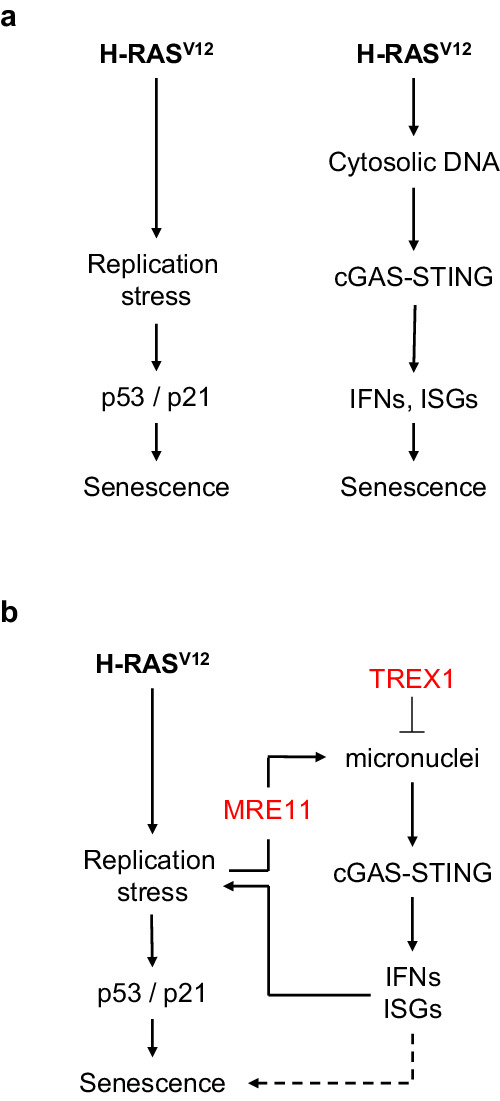
Oncogene-induced senescence (OIS) arrests cell proliferation in response to replication stress (RS) induced by oncogenes. OIS depends on the DNA damage response (DDR), but also on the cGAS-STING pathway, which detects cytosolic DNA and induces type I interferons (IFNs). Whether and how RS and IFN responses cooperate to promote OIS remains unknown. Here, we show that the induction of OIS by the H-RASV12 oncogene in immortalized human fibroblasts depends on the MRE11 nuclease. Indeed, treatment with the MRE11 inhibitor Mirin prevented RS, micronuclei formation and IFN response induced by RASV12. Overexpression of the cytosolic nuclease TREX1 also prevented OIS. Conversely, overexpression of a dominant negative mutant of TREX1 or treatment with IFN-β was sufficient to induce RS and DNA damage, independent of RASV12 induction. These data suggest that the IFN response acts as a positive feedback loop to amplify DDR in OIS through a process regulated by MRE11 and TREX1.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8:537–555. doi: 10.1158/2159-8290.CD-17-1461. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
