

Bridge RNAs direct programmable recombination of target and donor DNA
- PMID: 38926615
- PMCID: PMC11208160
- DOI: 10.1038/s41586-024-07552-4
Bridge RNAs direct programmable recombination of target and donor DNA
Abstract
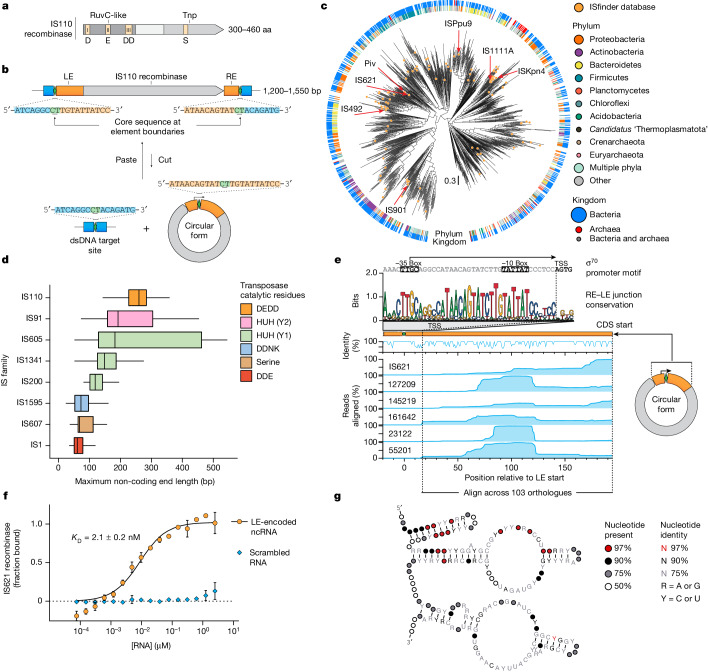
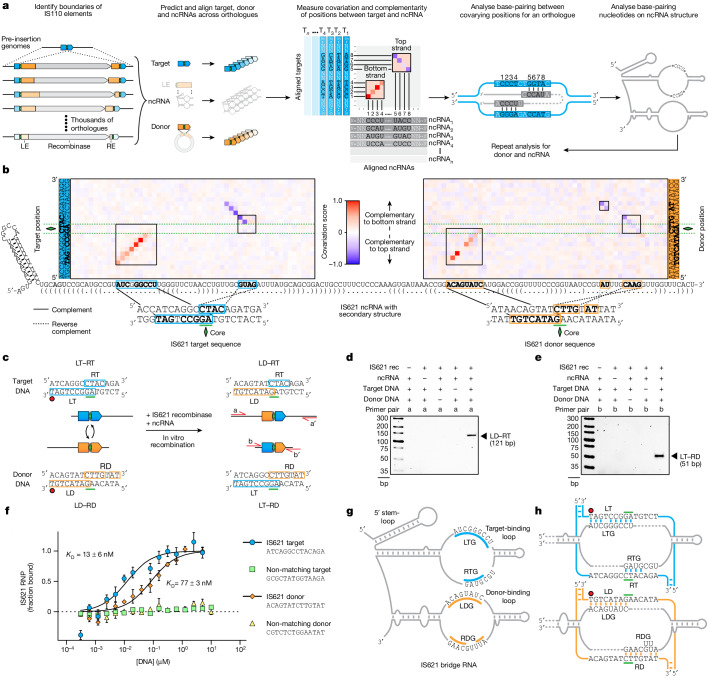
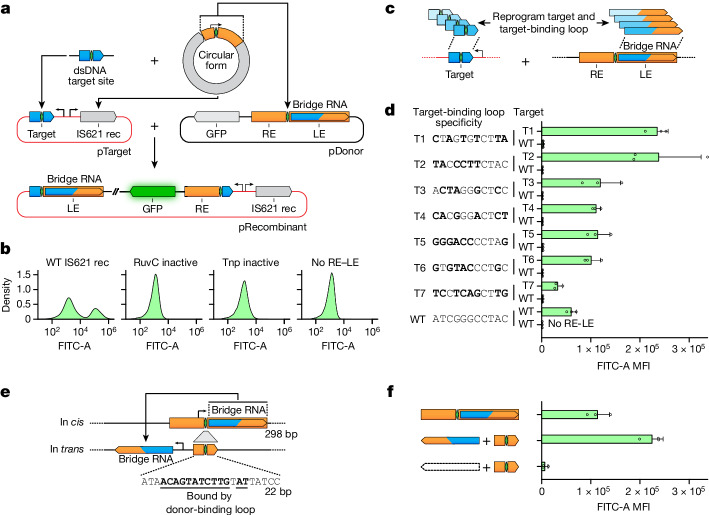
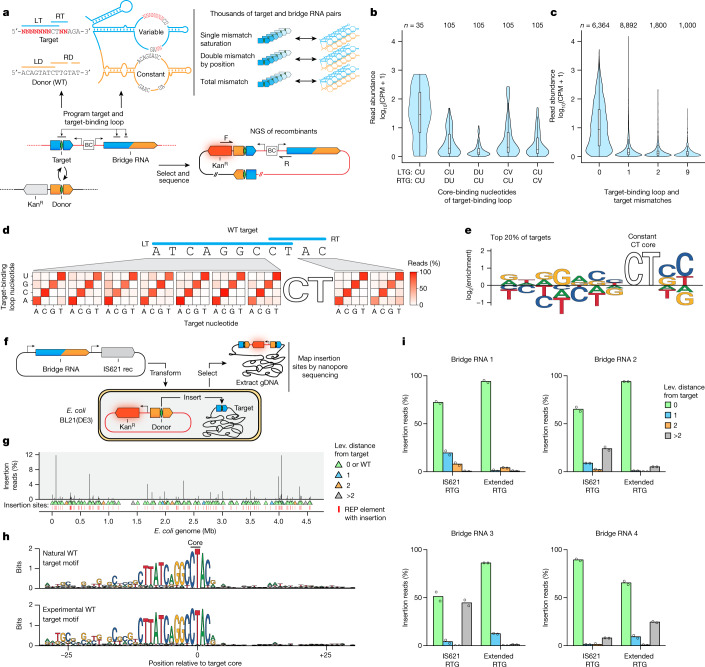
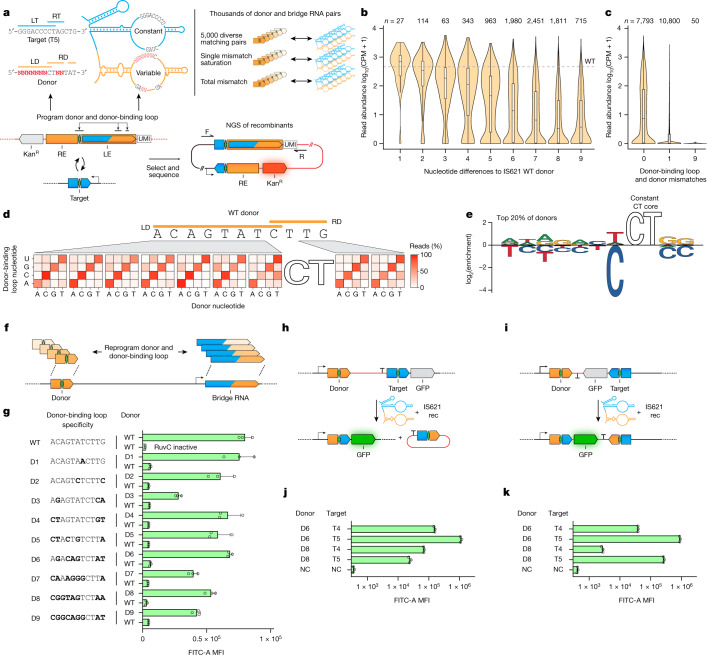
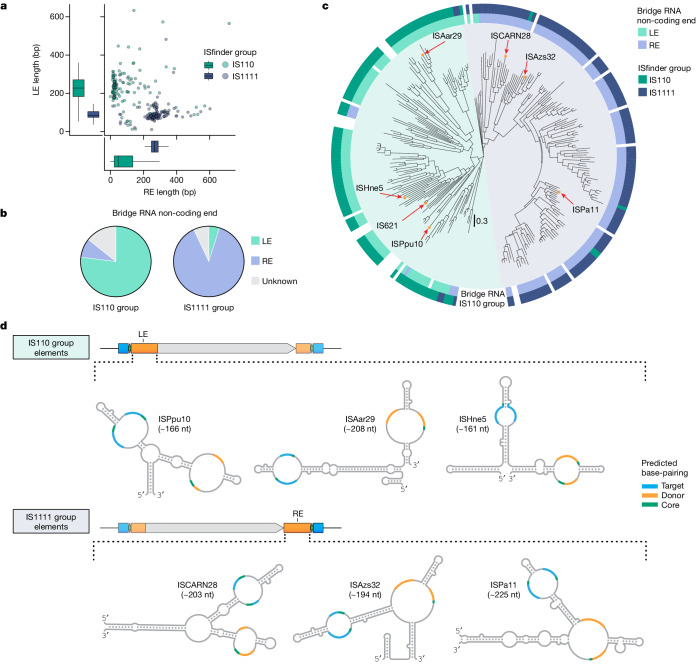
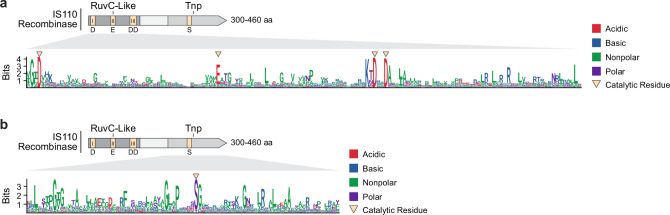
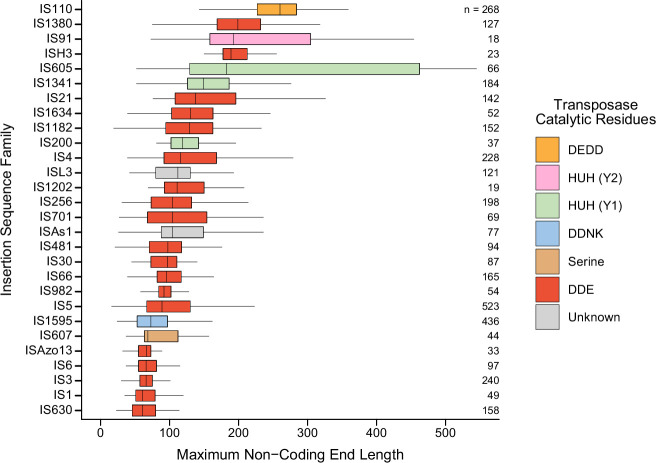
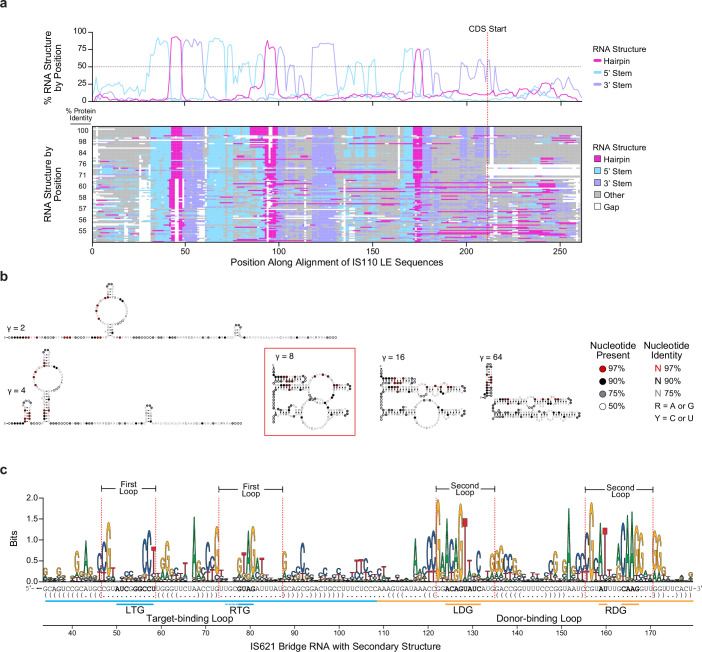
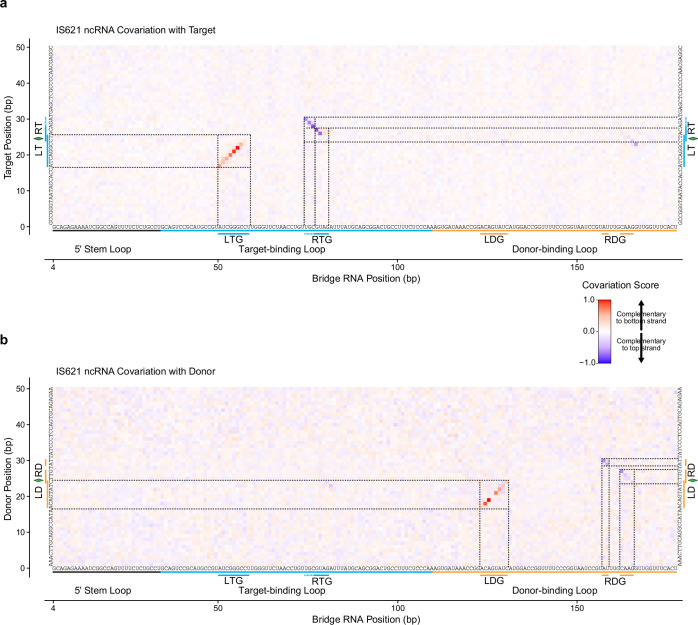
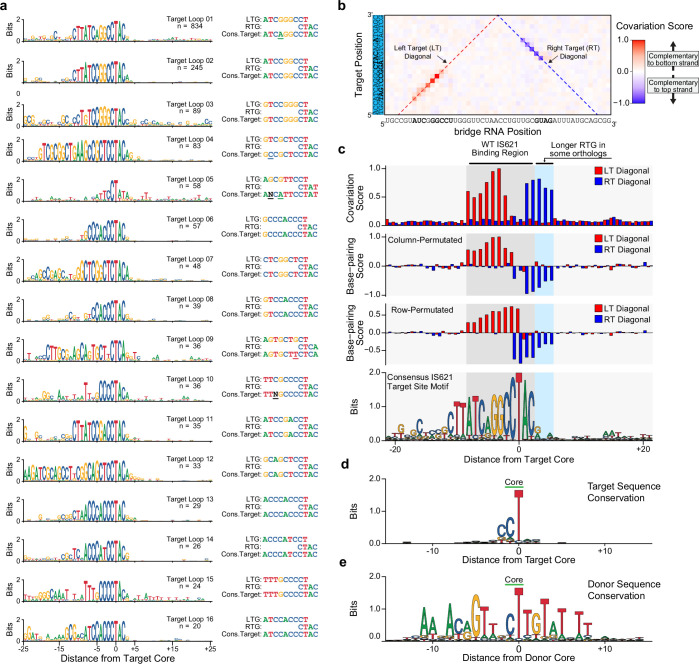
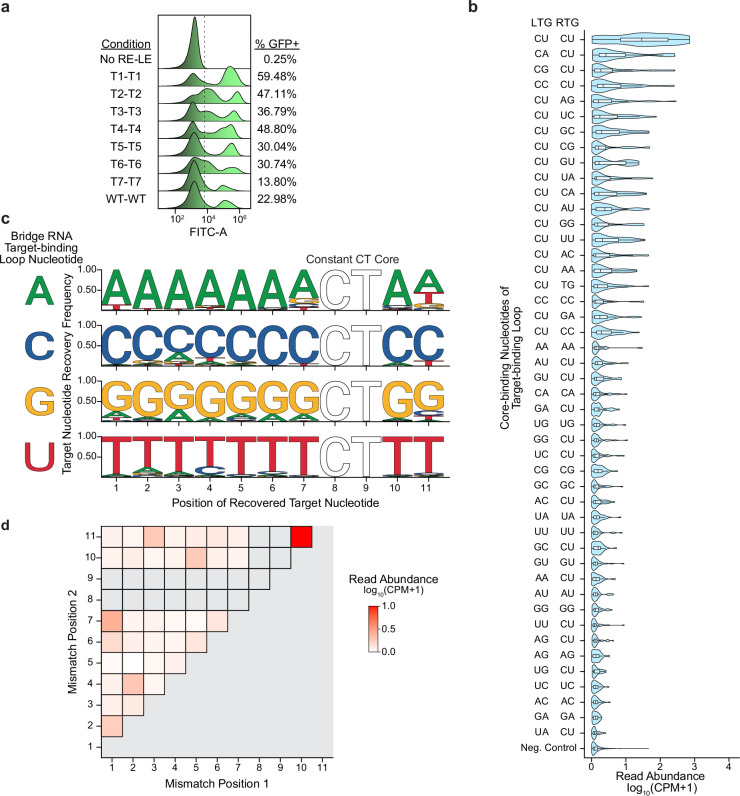
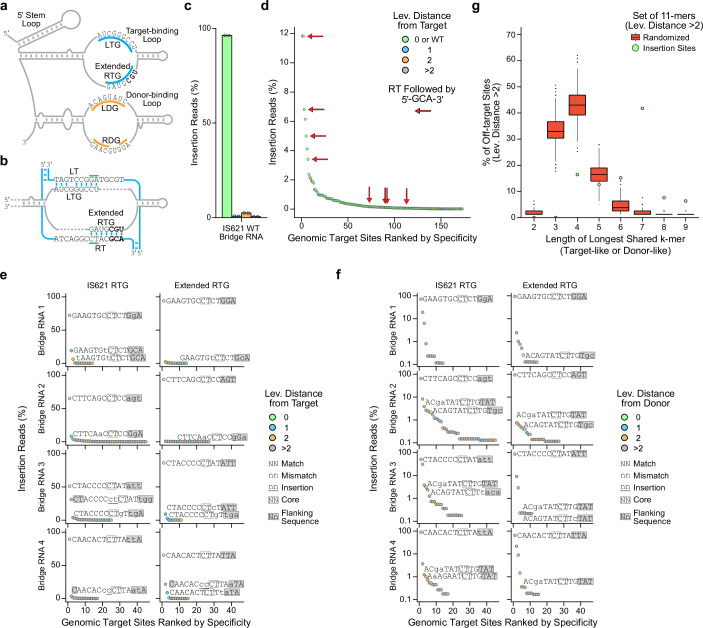
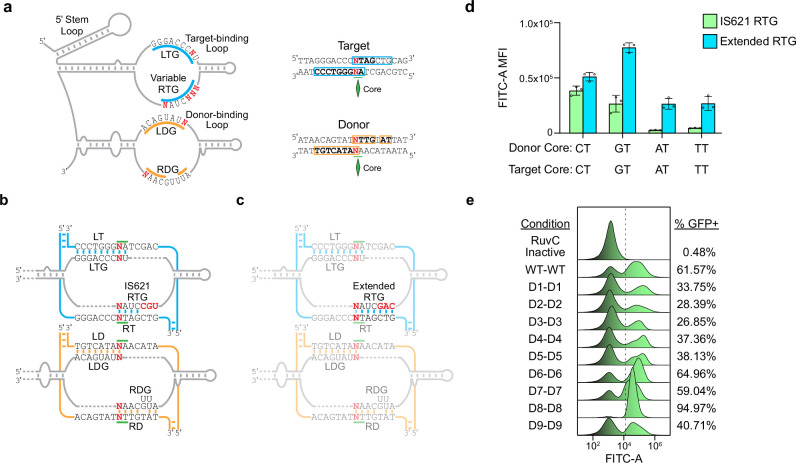
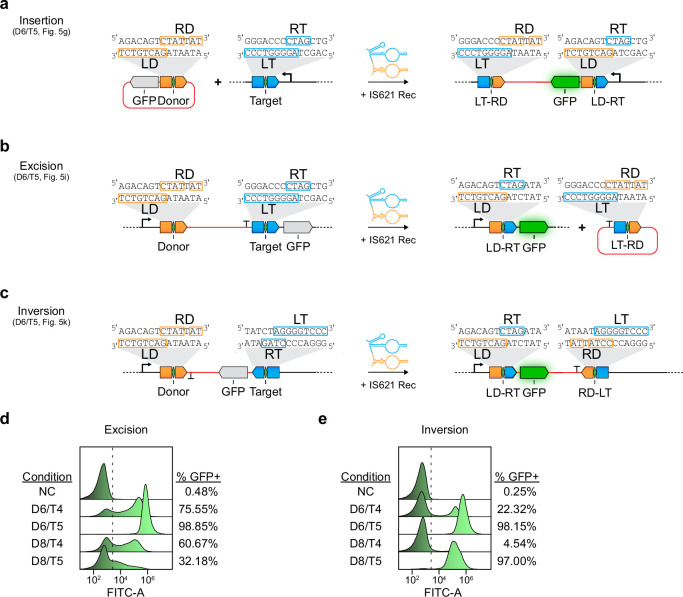
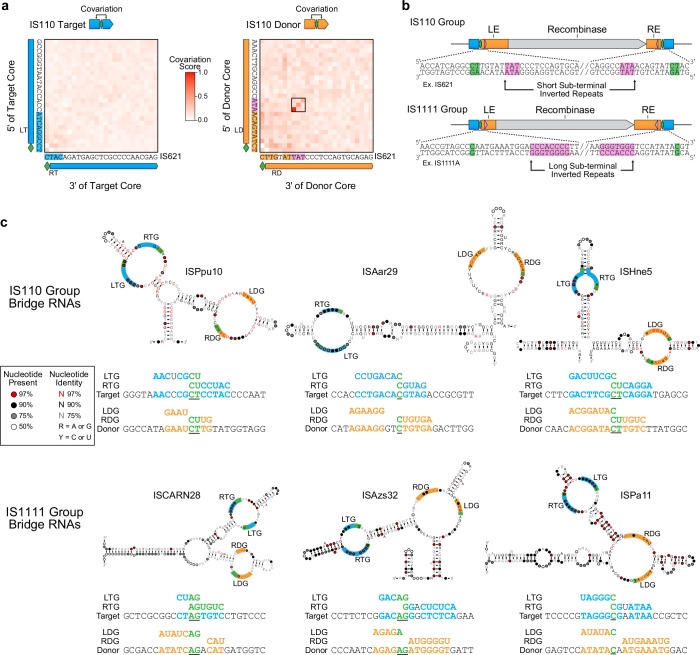
Genomic rearrangements, encompassing mutational changes in the genome such as insertions, deletions or inversions, are essential for genetic diversity. These rearrangements are typically orchestrated by enzymes that are involved in fundamental DNA repair processes, such as homologous recombination, or in the transposition of foreign genetic material by viruses and mobile genetic elements1,2. Here we report that IS110 insertion sequences, a family of minimal and autonomous mobile genetic elements, express a structured non-coding RNA that binds specifically to their encoded recombinase. This bridge RNA contains two internal loops encoding nucleotide stretches that base-pair with the target DNA and the donor DNA, which is the IS110 element itself. We demonstrate that the target-binding and donor-binding loops can be independently reprogrammed to direct sequence-specific recombination between two DNA molecules. This modularity enables the insertion of DNA into genomic target sites, as well as programmable DNA excision and inversion. The IS110 bridge recombination system expands the diversity of nucleic-acid-guided systems beyond CRISPR and RNA interference, offering a unified mechanism for the three fundamental DNA rearrangements-insertion, excision and inversion-that are required for genome design.
© 2024. The Author(s).
Conflict of interest statement
P.D.H. acknowledges outside interest in Stylus Medicine, Circle Labs, Spotlight Therapeutics, Arbor Biosciences, Varda Space, Vial Health and Veda Bio, in which he holds various roles including as co-founder, director, scientific advisory board member or consultant. M.G.D. acknowledges outside interest in Stylus Medicine. P.D.H., M.G.D., N.T.P., S.K., J.S.A., M.H. and H.N. are inventors on patents relating to this work. The remaining authors declare no competing interests.
Figures
Update of
-
Bridge RNAs direct modular and programmable recombination of target and donor DNA.bioRxiv [Preprint]. 2024 Jan 26:2024.01.24.577089. doi: 10.1101/2024.01.24.577089. bioRxiv. 2024. Update in: Nature. 2024 Jun;630(8018):984-993. doi: 10.1038/s41586-024-07552-4. PMID: 38328150 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
