

Inflammation-associated ectopic mineralization
- PMID: 38933004
- PMCID: PMC11197766
- DOI: 10.1016/j.fmre.2022.04.020
Inflammation-associated ectopic mineralization
Abstract
Ectopic mineralization refers to the deposition of mineralized complexes in the extracellular matrix of soft tissues. Calcific aortic valve disease, vascular calcification, gallstones, kidney stones, and abnormal mineralization in arthritis are common examples of ectopic mineralization. They are debilitating diseases and exhibit excess mortality, disability, and morbidity, which impose on patients with limited social or financial resources. Recent recognition that inflammation plays an important role in ectopic mineralization has attracted the attention of scientists from different research fields. In the present review, we summarize the origin of inflammation in ectopic mineralization and different channels whereby inflammation drives the initiation and progression of ectopic mineralization. The current knowledge of inflammatory milieu in pathological mineralization is reviewed, including how immune cells, pro-inflammatory mediators, and osteogenic signaling pathways induce the osteogenic transition of connective tissue cells, providing nucleating sites and assembly of aberrant minerals. Advances in the understanding of the underlying mechanisms involved in inflammatory-mediated ectopic mineralization enable novel strategies to be developed that may lead to the resolution of these enervating conditions.
Keywords: Anti-inflammatory treatments; Ectopic mineralization; Immune cells; Inflammatory conditions; Inflammatory mediators; Osteogenic signaling pathways.
© 2022 The Authors. Publishing Services by Elsevier B.V. on behalf of KeAi Communications Co. Ltd.
Conflict of interest statement
The authors declare that they have no conflicts of interest in this work.
Figures



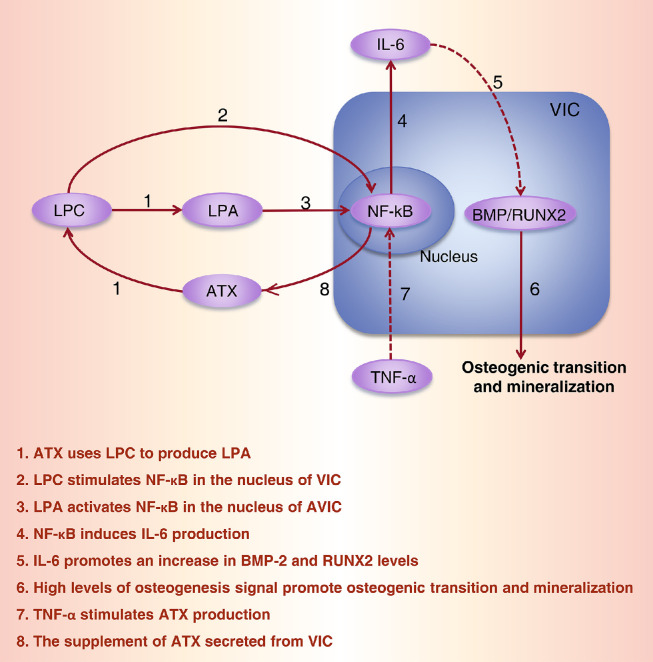


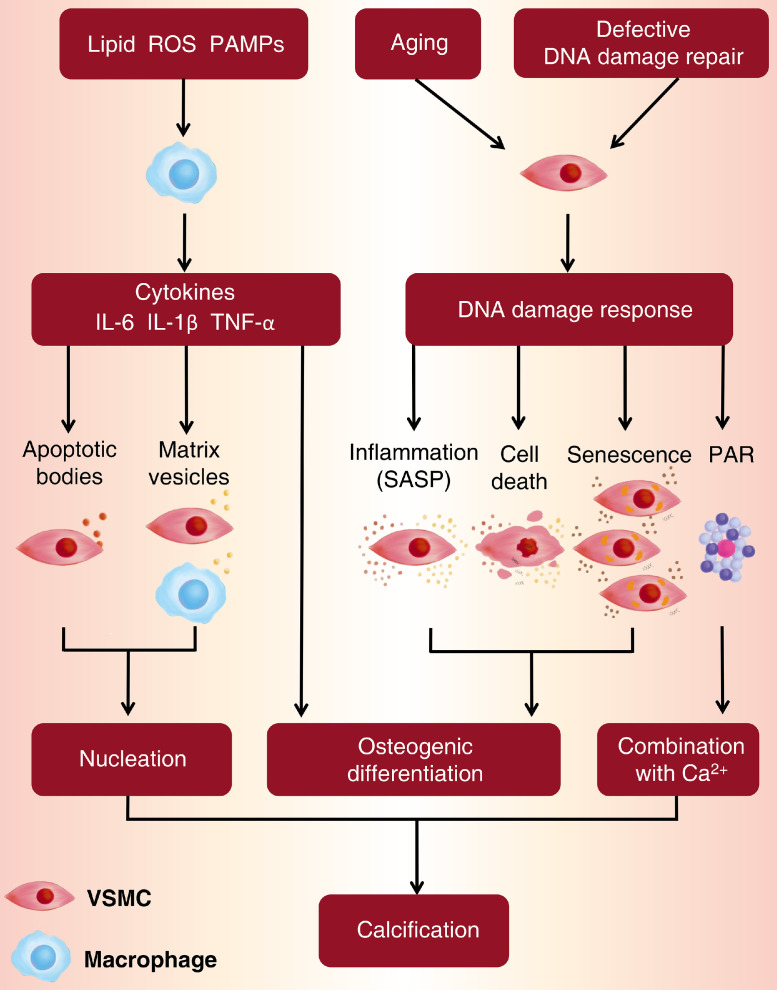


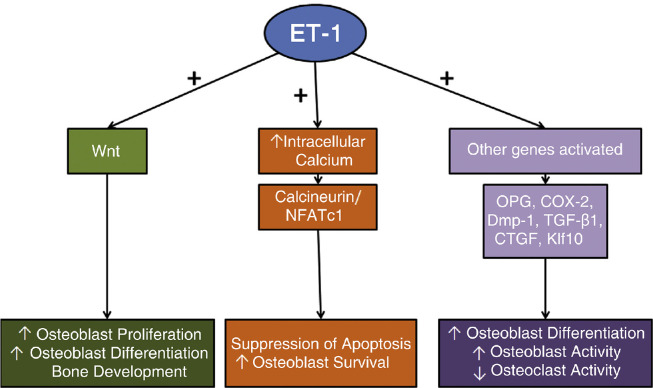
References
-
- Cherng J.H., Hsu Y.J., Liu C.C., et al. Activities of Ca2+-related ion channels during the formation of kidney stones in an infection-induced urolithiasis rat model. Am. J. Physiol. Ren. Physiol. 2019;317(5):F1342–F1349. - PubMed
-
- Parente F., Pastore L., Bargiggia S., et al. Incidence and risk factors for gallstones in patients with inflammatory bowel disease: a large case-control study. Hepatology. 2007;45(5):1267–1274. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
