

CapTrap-seq: a platform-agnostic and quantitative approach for high-fidelity full-length RNA sequencing
- PMID: 38937428
- PMCID: PMC11211341
- DOI: 10.1038/s41467-024-49523-3
CapTrap-seq: a platform-agnostic and quantitative approach for high-fidelity full-length RNA sequencing
Abstract
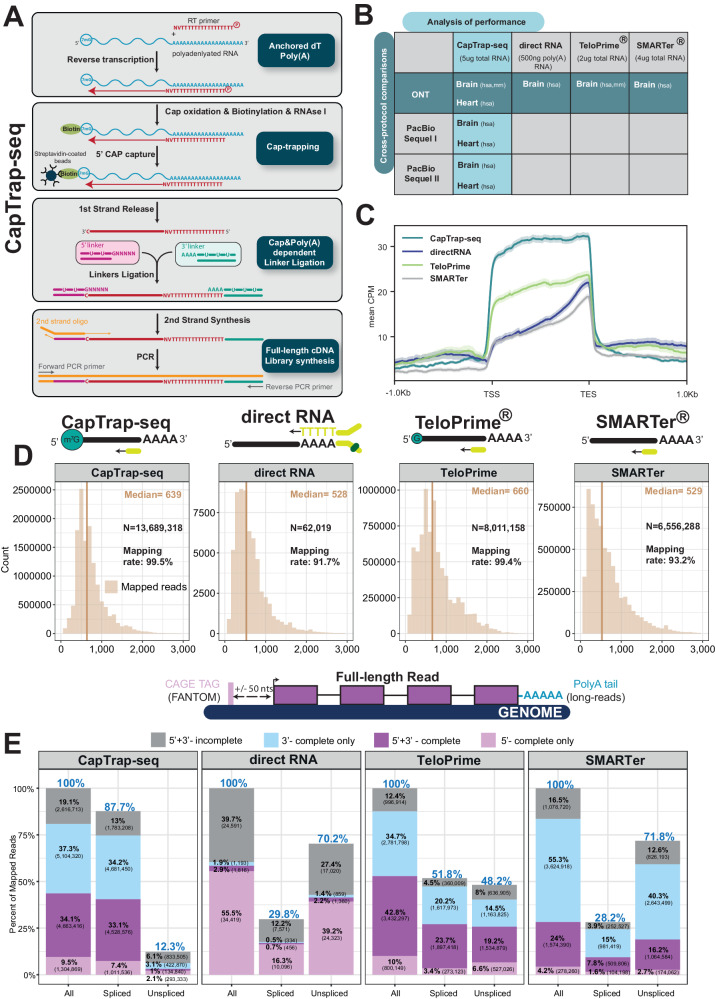
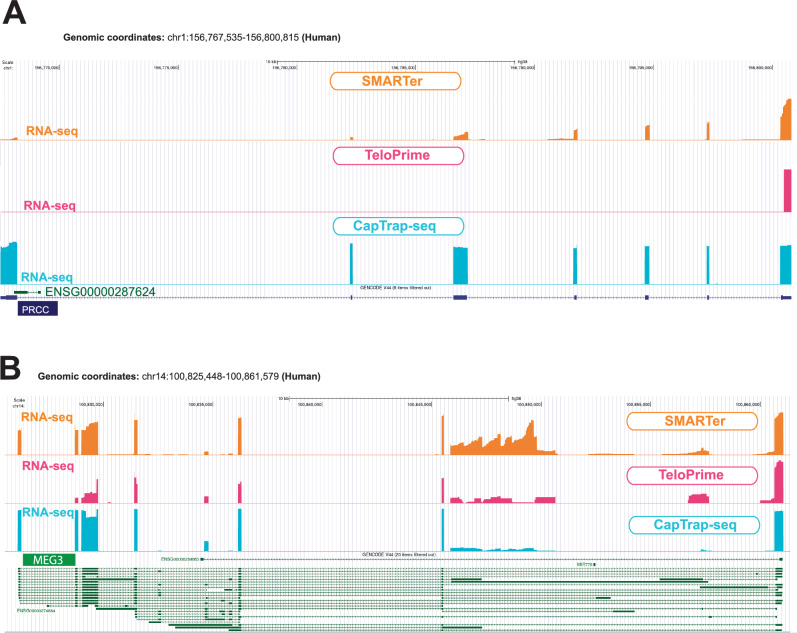
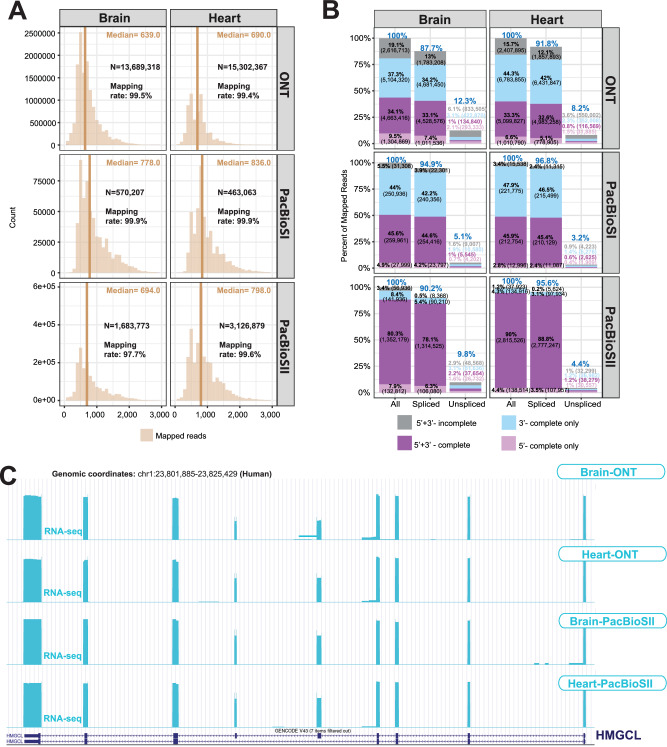
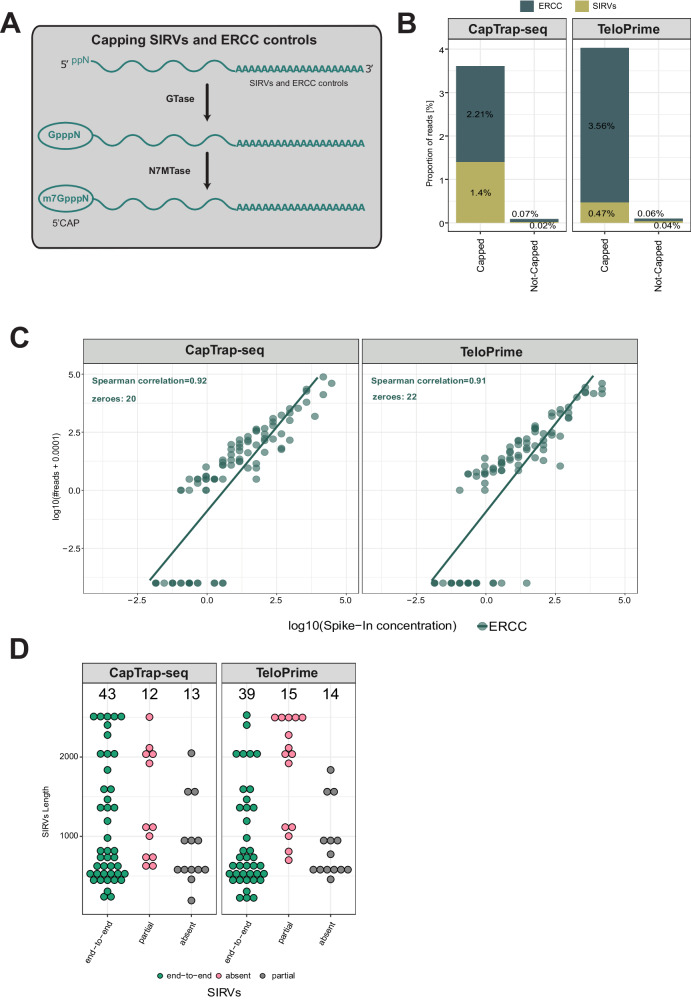
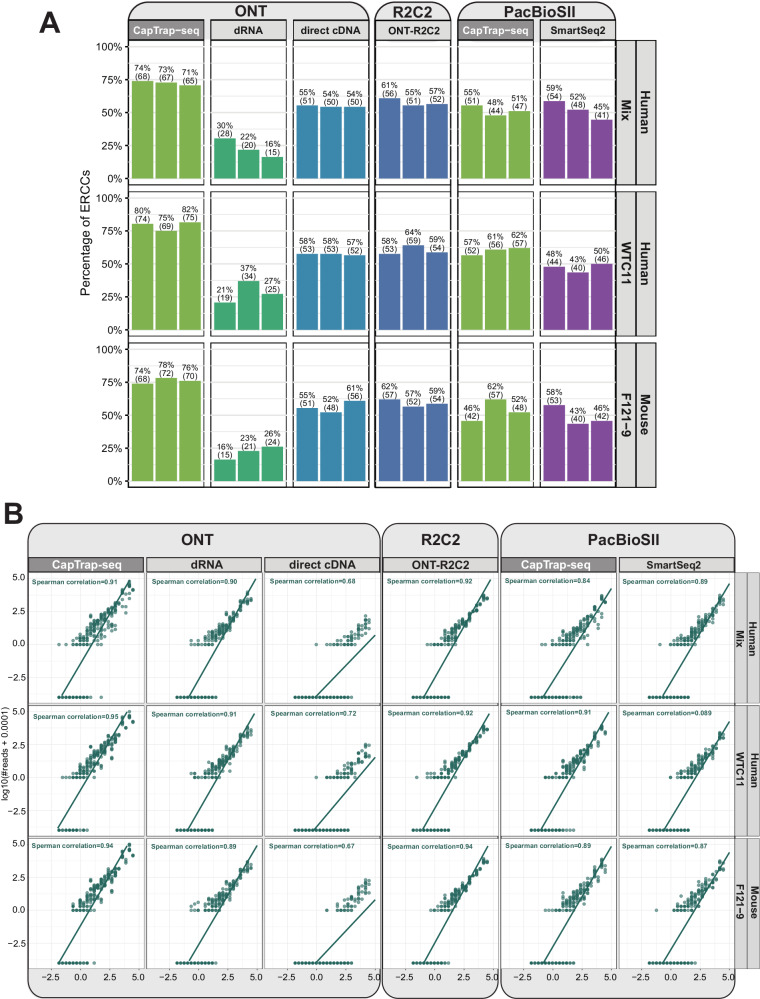
Long-read RNA sequencing is essential to produce accurate and exhaustive annotation of eukaryotic genomes. Despite advancements in throughput and accuracy, achieving reliable end-to-end identification of RNA transcripts remains a challenge for long-read sequencing methods. To address this limitation, we develop CapTrap-seq, a cDNA library preparation method, which combines the Cap-trapping strategy with oligo(dT) priming to detect 5' capped, full-length transcripts. In our study, we evaluate the performance of CapTrap-seq alongside other widely used RNA-seq library preparation protocols in human and mouse tissues, employing both ONT and PacBio sequencing technologies. To explore the quantitative capabilities of CapTrap-seq and its accuracy in reconstructing full-length RNA molecules, we implement a capping strategy for synthetic RNA spike-in sequences that mimics the natural 5'cap formation. Our benchmarks, incorporating the Long-read RNA-seq Genome Annotation Assessment Project (LRGASP) data, demonstrate that CapTrap-seq is a competitive, platform-agnostic RNA library preparation method for generating full-length transcript sequences.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
CapTrap-Seq: A platform-agnostic and quantitative approach for high-fidelity full-length RNA transcript sequencing.bioRxiv [Preprint]. 2023 Jun 18:2023.06.16.543444. doi: 10.1101/2023.06.16.543444. bioRxiv. 2023. Update in: Nat Commun. 2024 Jun 27;15(1):5278. doi: 10.1038/s41467-024-49523-3. PMID: 37398314 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
