

SpecEncoder: deep metric learning for accurate peptide identification in proteomics
- PMID: 38940141
- PMCID: PMC11211836
- DOI: 10.1093/bioinformatics/btae220
SpecEncoder: deep metric learning for accurate peptide identification in proteomics
Abstract
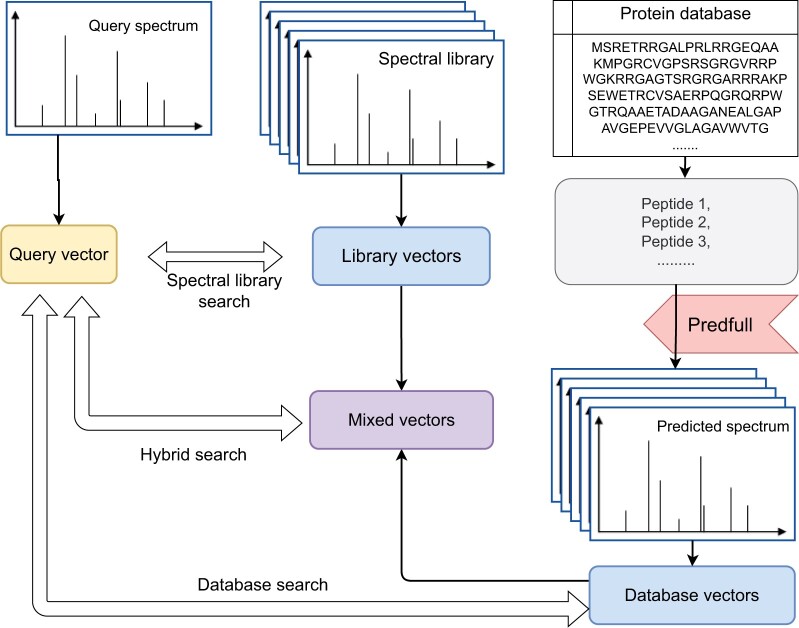
Motivation: Tandem mass spectrometry (MS/MS) is a crucial technology for large-scale proteomic analysis. The protein database search or the spectral library search are commonly used for peptide identification from MS/MS spectra, which, however, may face challenges due to experimental variations between replicated spectra and similar fragmentation patterns among distinct peptides. To address this challenge, we present SpecEncoder, a deep metric learning approach to address these challenges by transforming MS/MS spectra into robust and sensitive embedding vectors in a latent space. The SpecEncoder model can also embed predicted MS/MS spectra of peptides, enabling a hybrid search approach that combines spectral library and protein database searches for peptide identification.
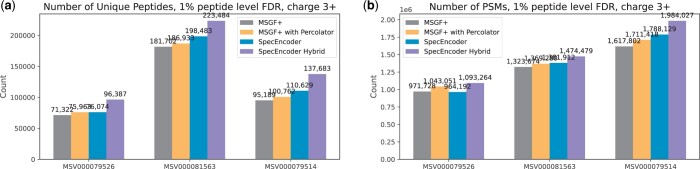
Results: We evaluated SpecEncoder on three large human proteomics datasets, and the results showed a consistent improvement in peptide identification. For spectral library search, SpecEncoder identifies 1%-2% more unique peptides (and PSMs) than SpectraST. For protein database search, it identifies 6%-15% more unique peptides than MSGF+ enhanced by Percolator, Furthermore, SpecEncoder identified 6%-12% additional unique peptides when utilizing a combined library of experimental and predicted spectra. SpecEncoder can also identify more peptides when compared to deep-learning enhanced methods (MSFragger boosted by MSBooster). These results demonstrate SpecEncoder's potential to enhance peptide identification for proteomic data analyses.
Availability and implementation: The source code and scripts for SpecEncoder and peptide identification are available on GitHub at https://github.com/lkytal/SpecEncoder. Contact: hatang@iu.edu.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures





References
-
- Aebersold R, Mann M.. Mass spectrometry-based proteomics. Nature 2003;422:198–207. - PubMed
-
- Bai S, Kolter JZ, Koltun V. An empirical evaluation of generic convolutional and recurrent networks for sequence modeling. In: Proceeding of the Seventh International Conference on Learning Representations (ICLR), New Orleans, USA, May 6–May 9, 2019.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
