

The proteogenomic landscape of multiple myeloma reveals insights into disease biology and therapeutic opportunities
- PMID: 38942927
- PMCID: PMC11358022
- DOI: 10.1038/s43018-024-00784-3
The proteogenomic landscape of multiple myeloma reveals insights into disease biology and therapeutic opportunities
Abstract
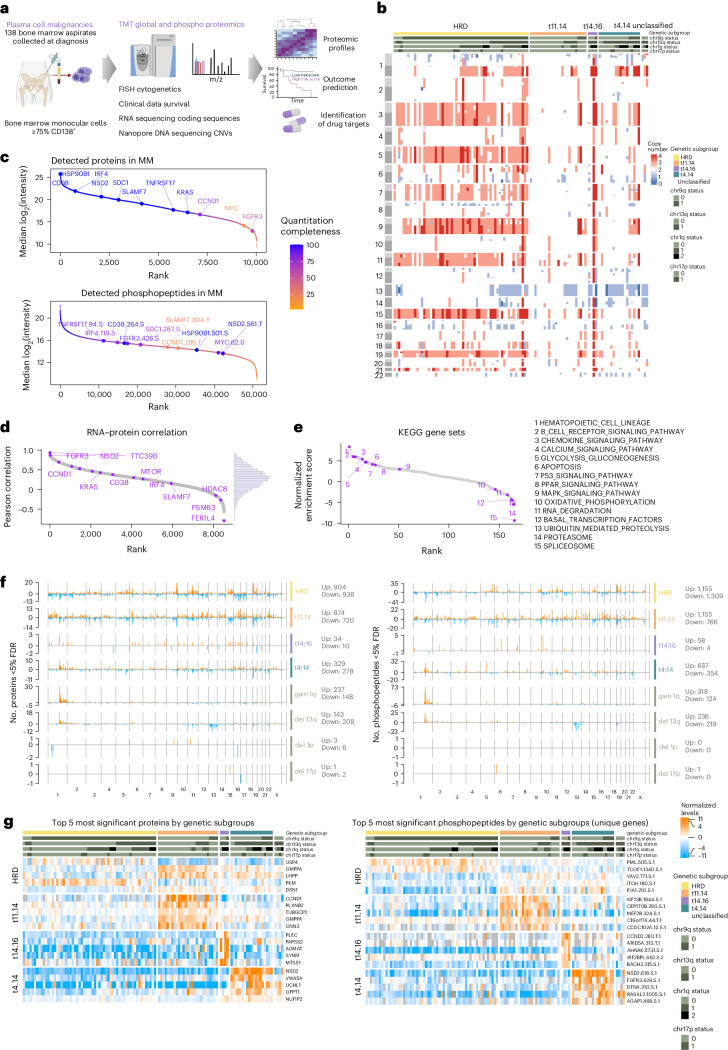
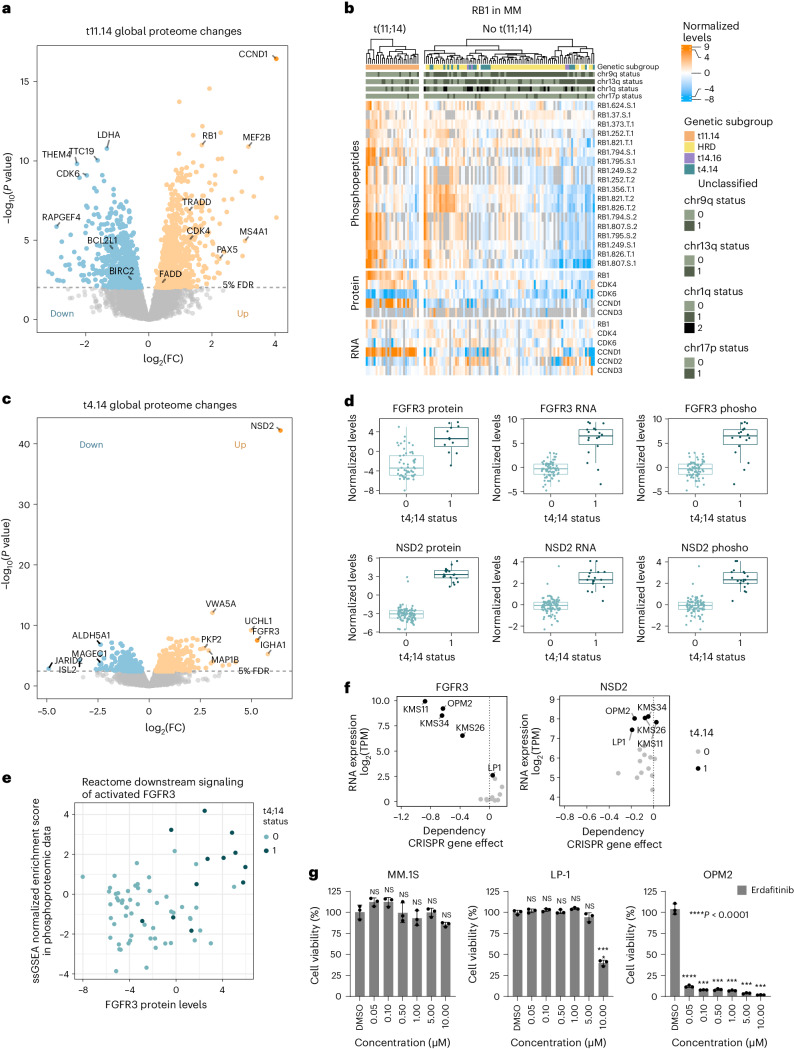
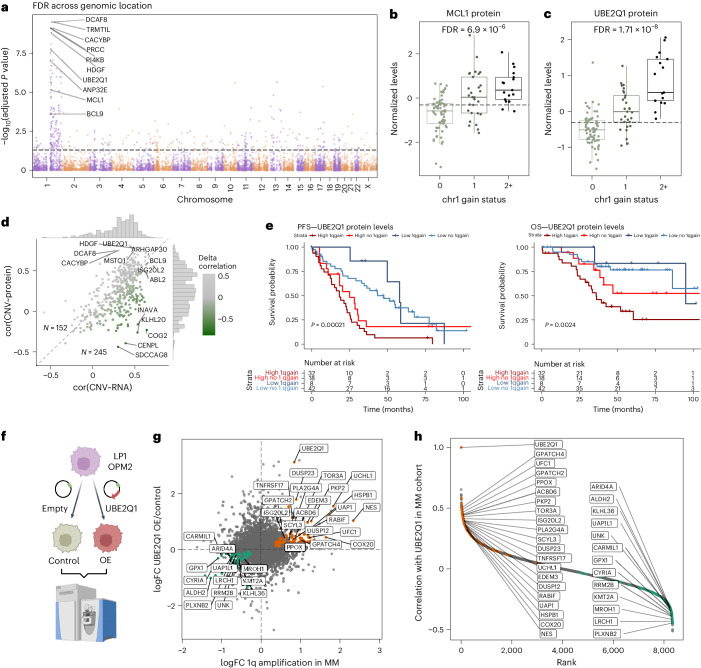
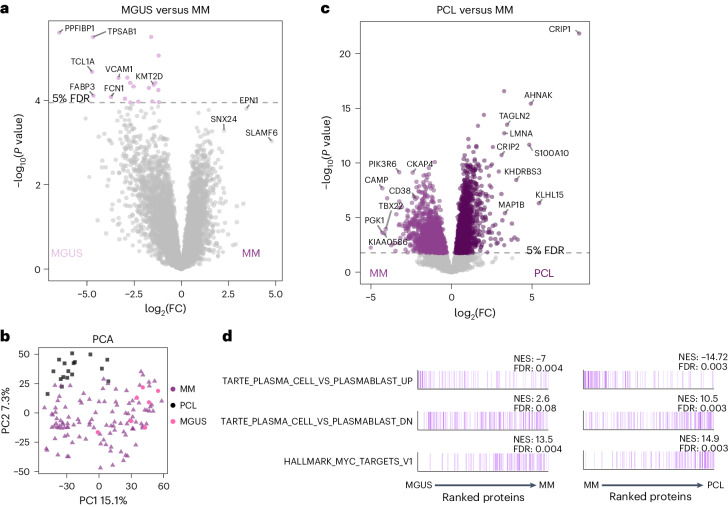
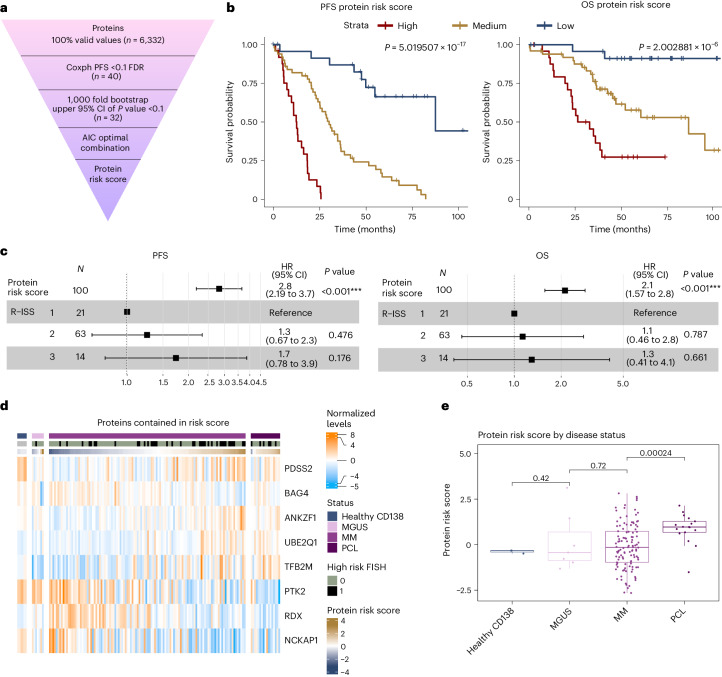
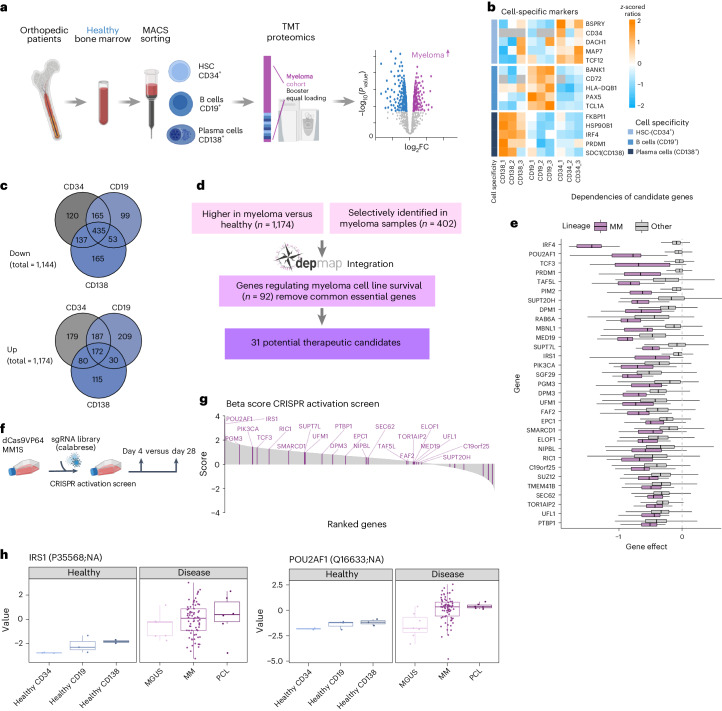
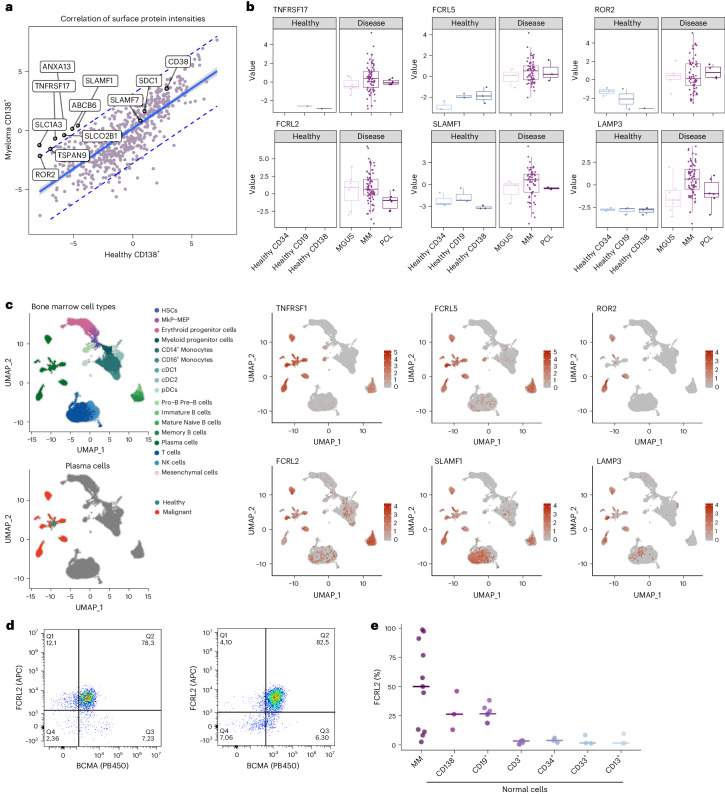
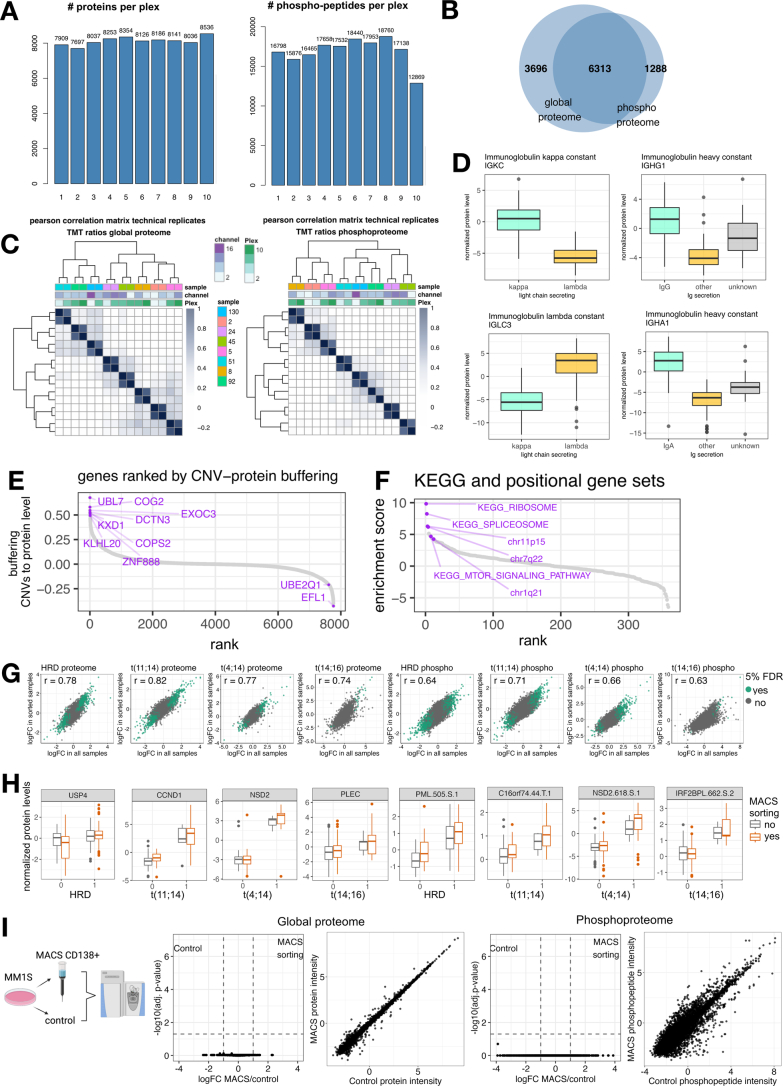
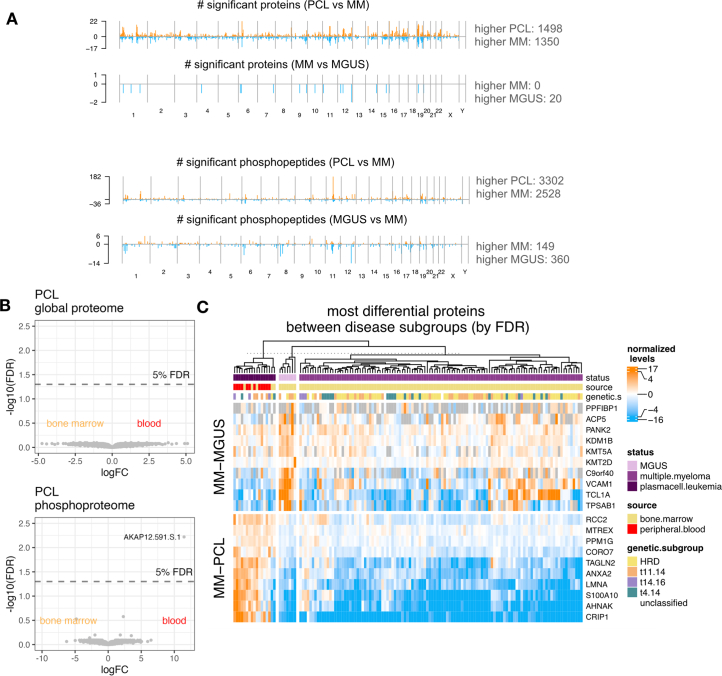
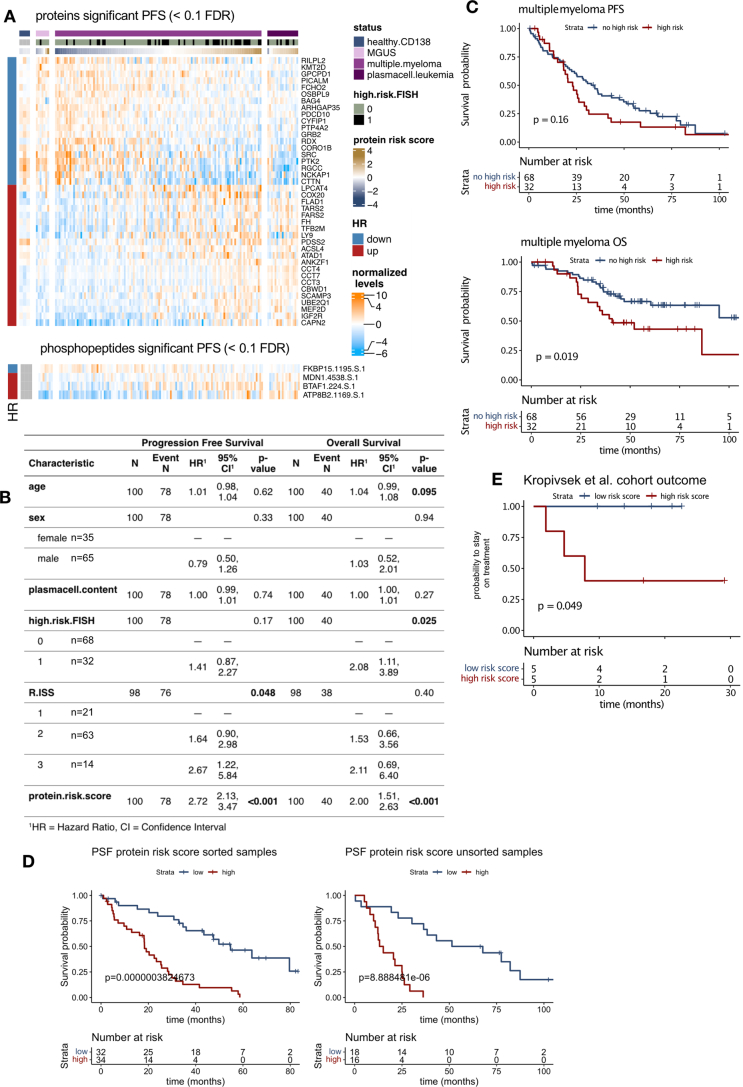
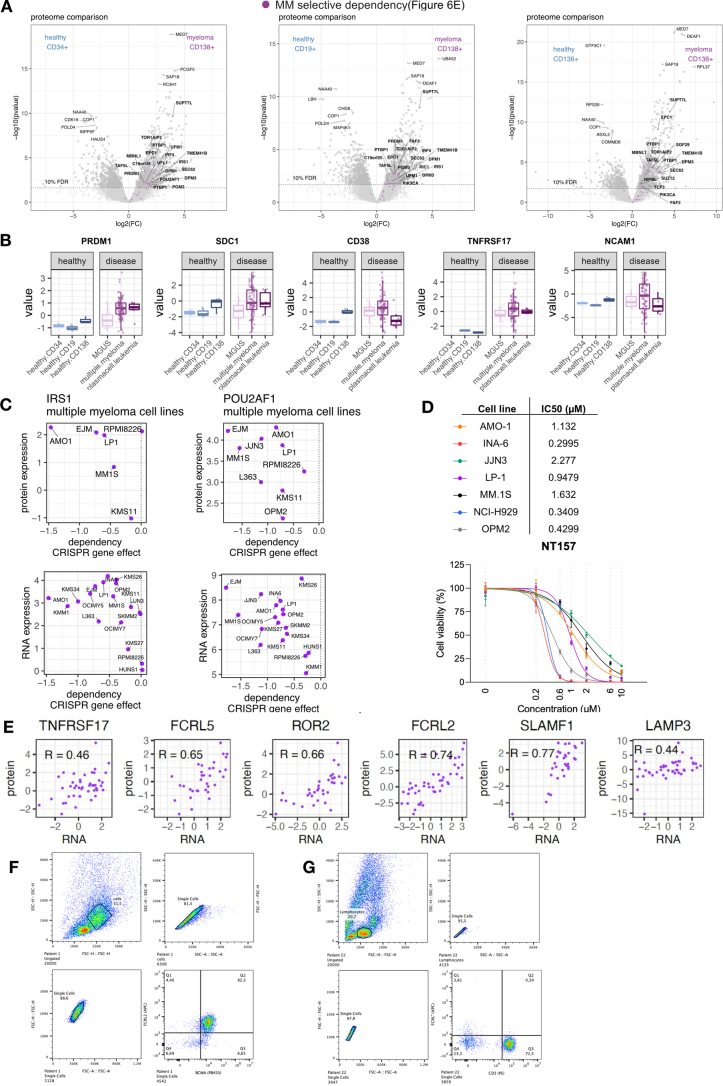
Multiple myeloma (MM) is a plasma cell malignancy of the bone marrow. Despite therapeutic advances, MM remains incurable, and better risk stratification as well as new therapies are therefore highly needed. The proteome of MM has not been systematically assessed before and holds the potential to uncover insight into disease biology and improved prognostication in addition to genetic and transcriptomic studies. Here we provide a comprehensive multiomics analysis including deep tandem mass tag-based quantitative global (phospho)proteomics, RNA sequencing, and nanopore DNA sequencing of 138 primary patient-derived plasma cell malignancies encompassing treatment-naive MM, plasma cell leukemia and the premalignancy monoclonal gammopathy of undetermined significance, as well as healthy controls. We found that the (phospho)proteome of malignant plasma cells are highly deregulated as compared with healthy plasma cells and is both defined by chromosomal alterations as well as posttranscriptional regulation. A prognostic protein signature was identified that is associated with aggressive disease independent of established risk factors in MM. Integration with functional genetics and single-cell RNA sequencing revealed general and genetic subtype-specific deregulated proteins and pathways in plasma cell malignancies that include potential targets for (immuno)therapies. Our study demonstrates the potential of proteogenomics in cancer and provides an easily accessible resource for investigating protein regulation and new therapeutic approaches in MM.
© 2024. The Author(s).
Conflict of interest statement
J.K. received speaker and/or advisory board honoraria from Bristol-Myers Squibb/Celgene, Sanofi, Abbvie, Takeda, Pfizer and Janssen. S.K. received honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, Takeda, Sanofi and Oncopeptides; served as a consultant or in an advisory role for Amgen, Bristol-Myers Squibb, Celgene, Janssen and Takeda; and received research funding from Amgen, Bristol-Myers Squibb, Celgene, Janssen and Takeda. The other authors declare no competing interests.
Figures

References
-
- van de Donk, N. W. C. J., Pawlyn, C. & Yong, K. L. Multiple myeloma. Lancet397, 410–427 (2021). - PubMed
-
- Manier, S. et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol.14, 100–113 (2017). - PubMed
-
- Shaughnessy, J. D. Jr et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood109, 2276–2284 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
