

The genetic architecture of biological age in nine human organ systems
- PMID: 38942983
- PMCID: PMC11446180
- DOI: 10.1038/s43587-024-00662-8
The genetic architecture of biological age in nine human organ systems
Abstract
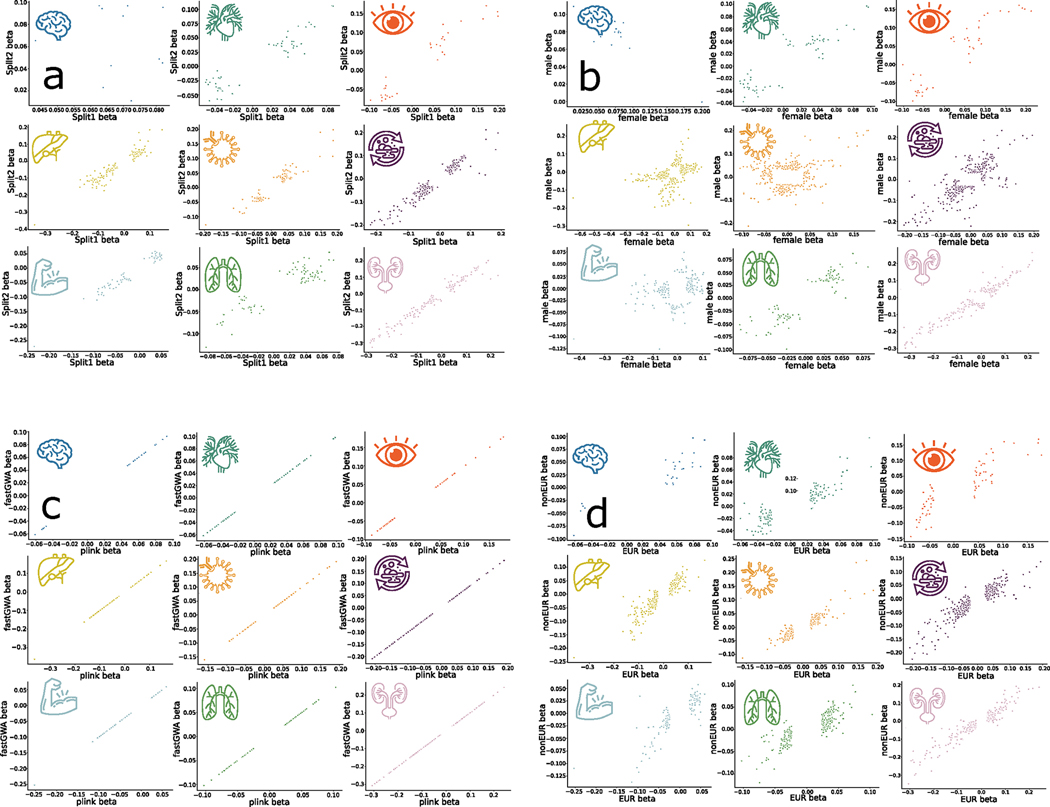
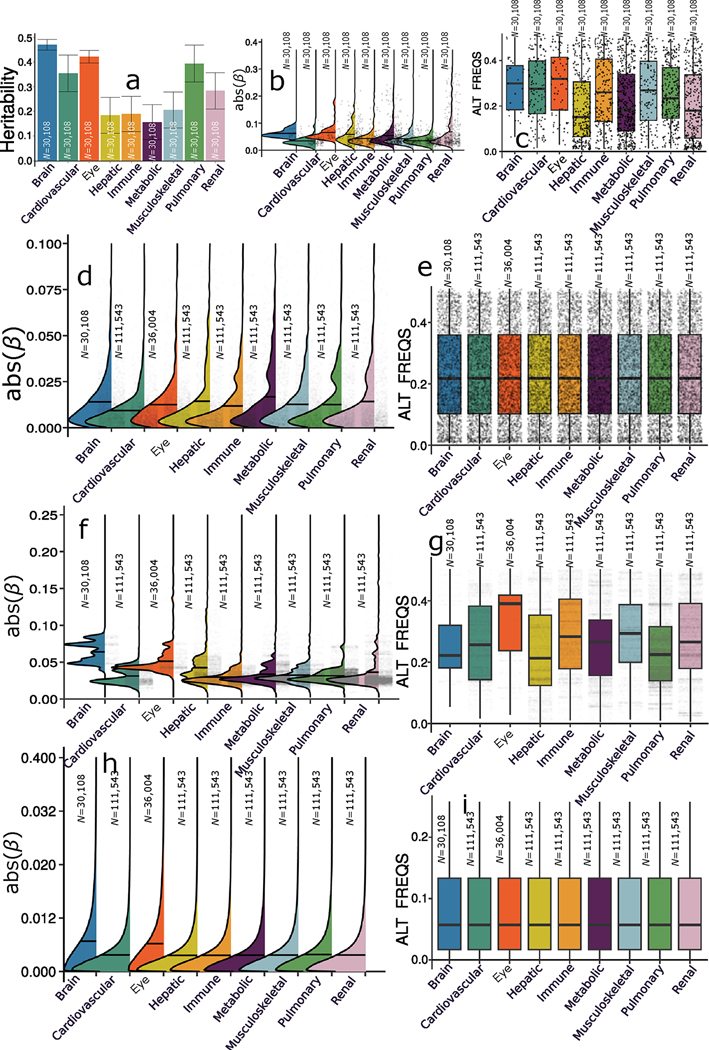
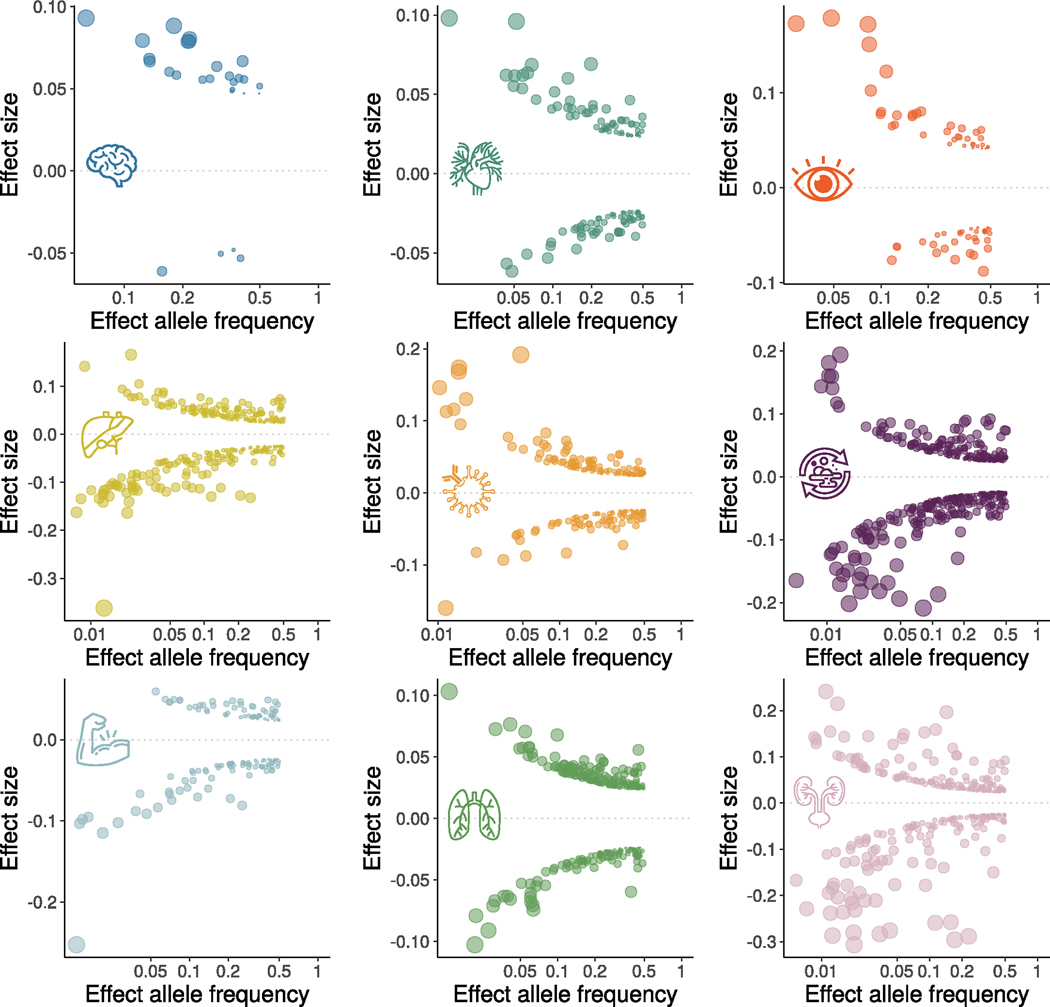
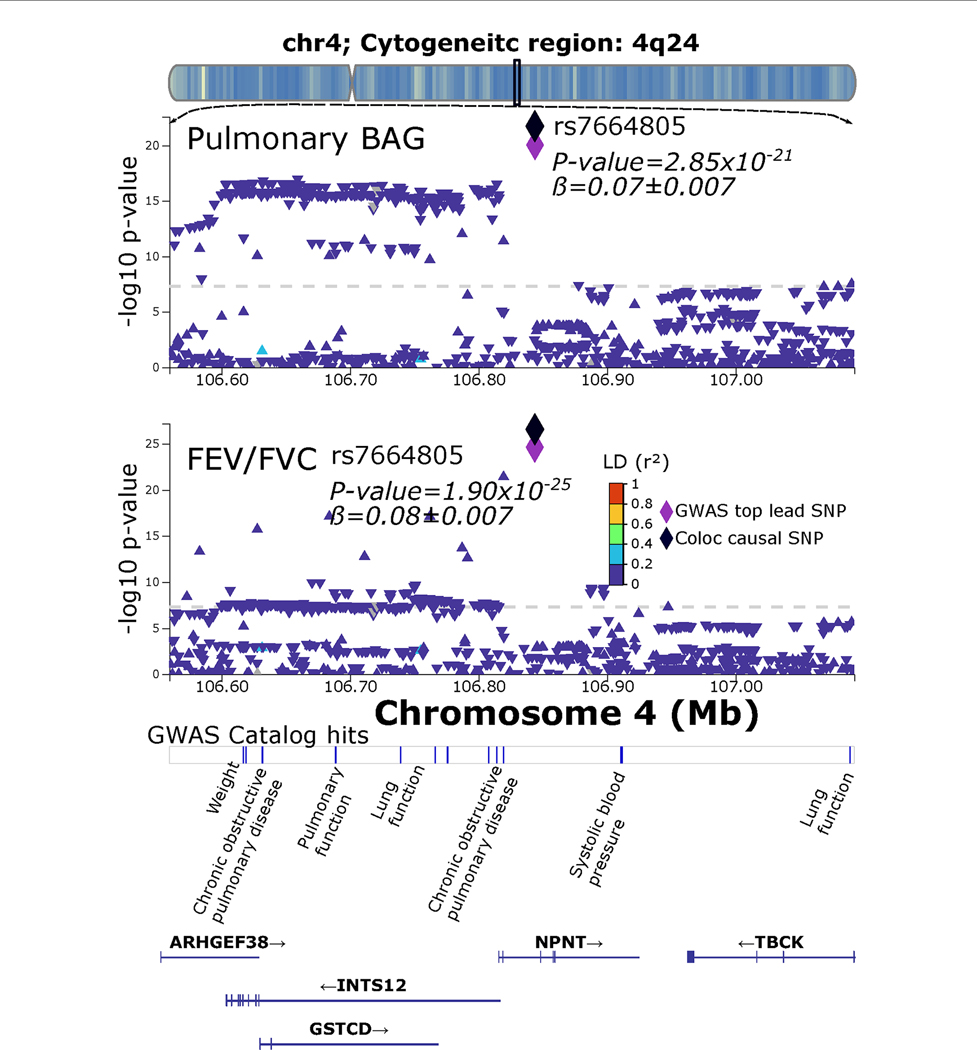
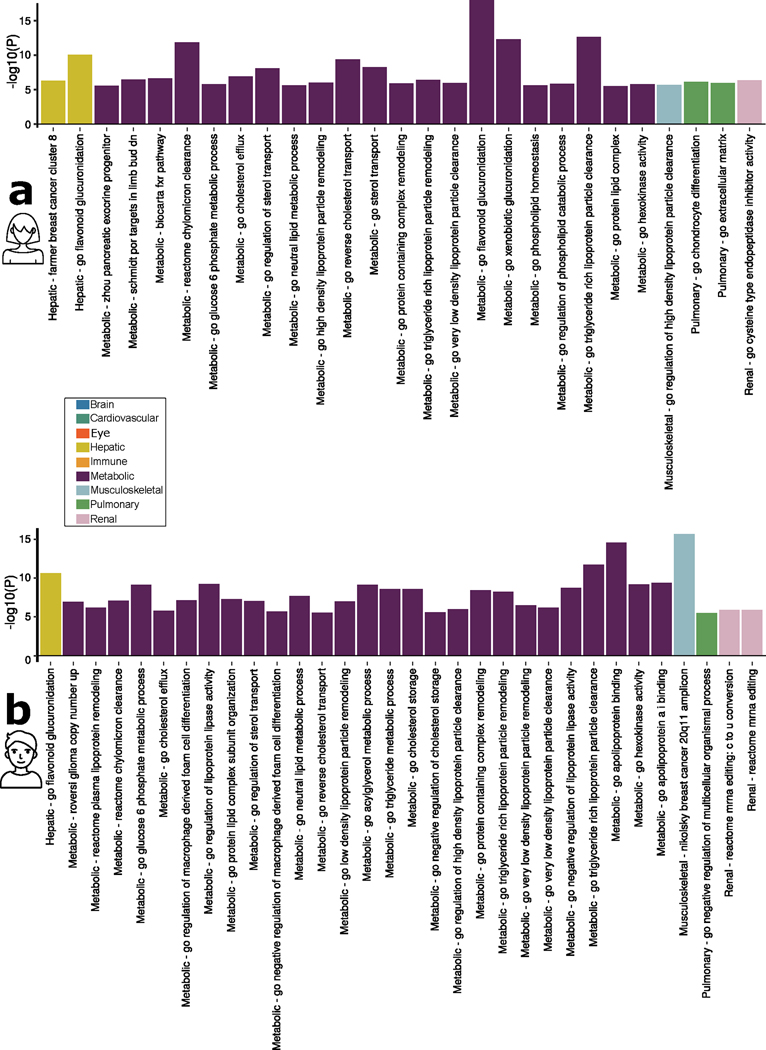
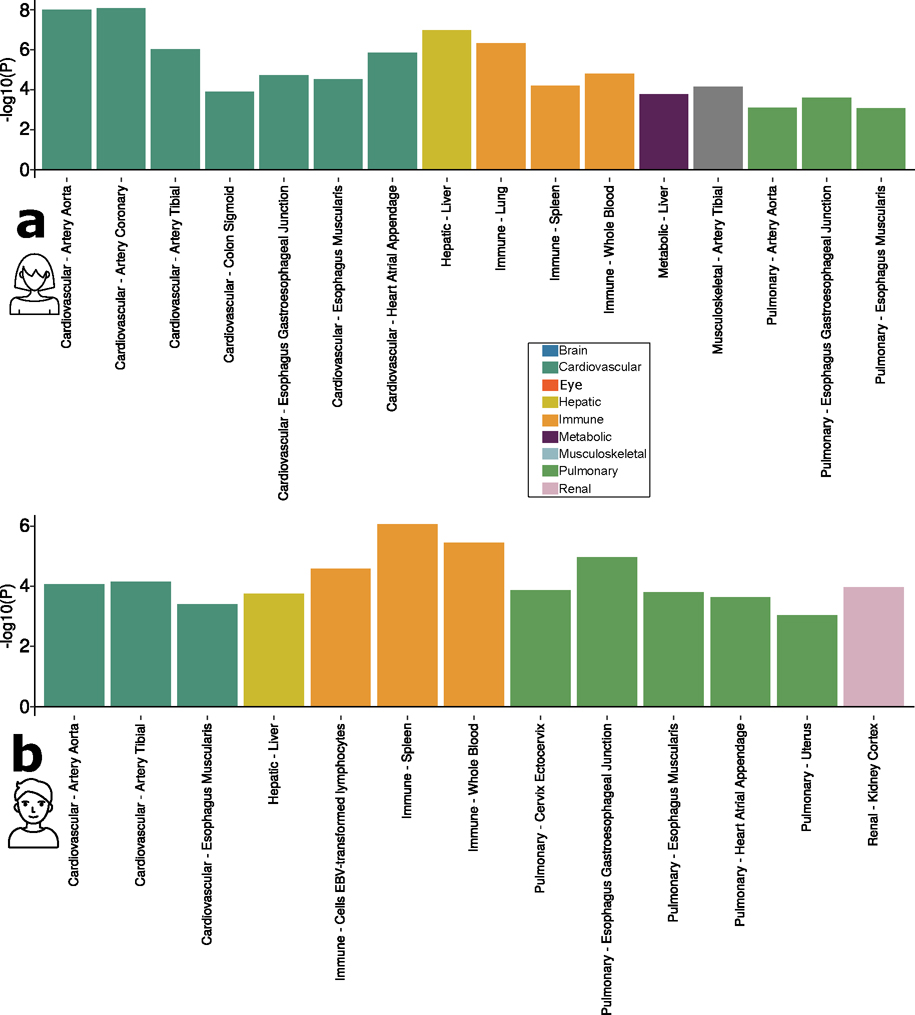
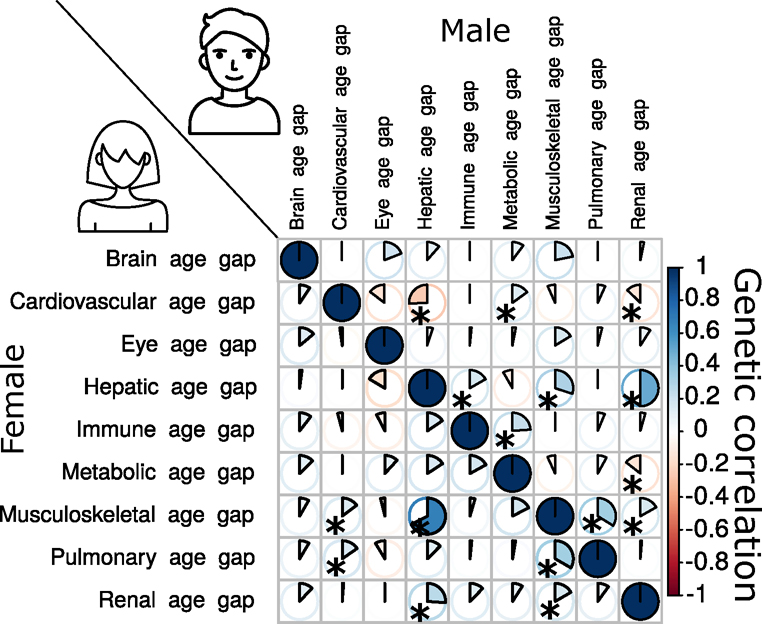
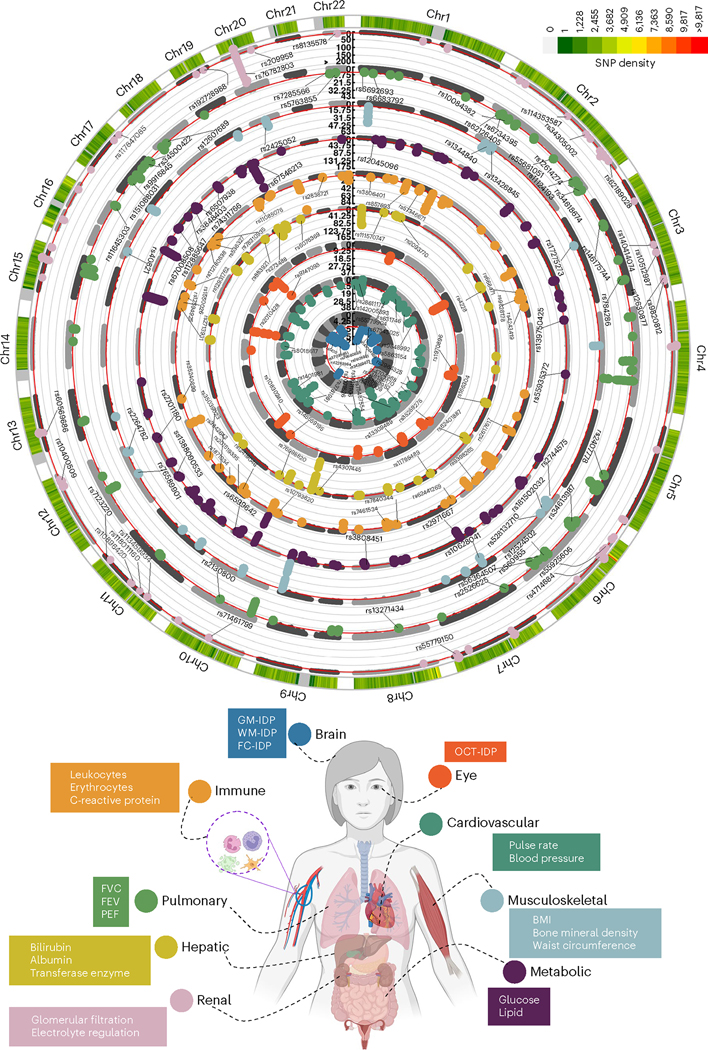
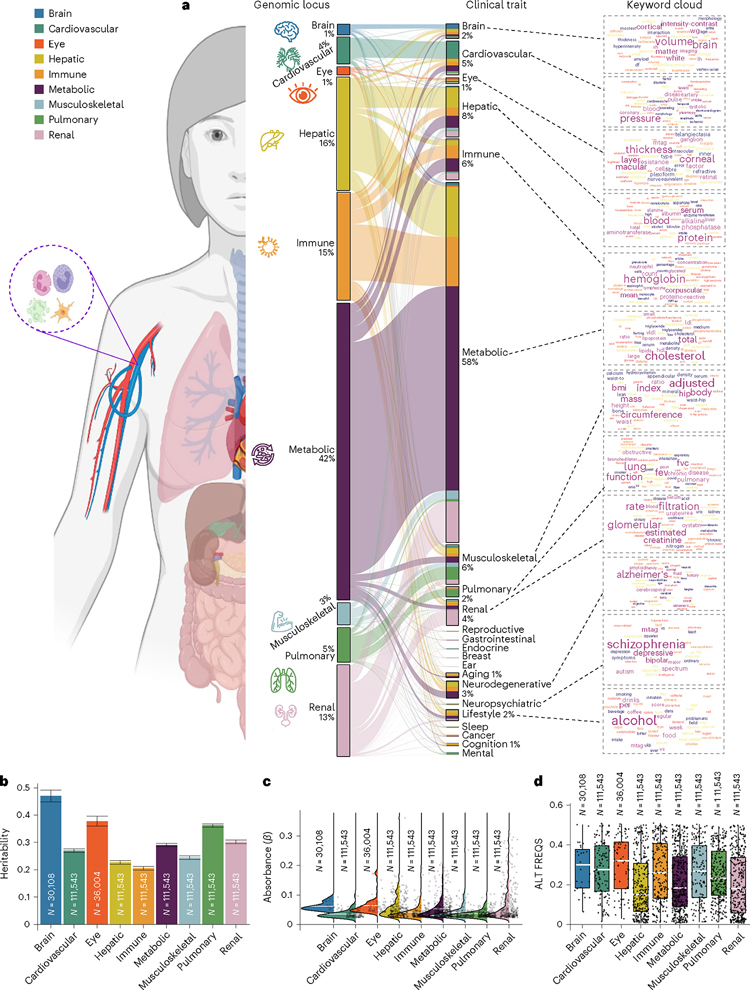
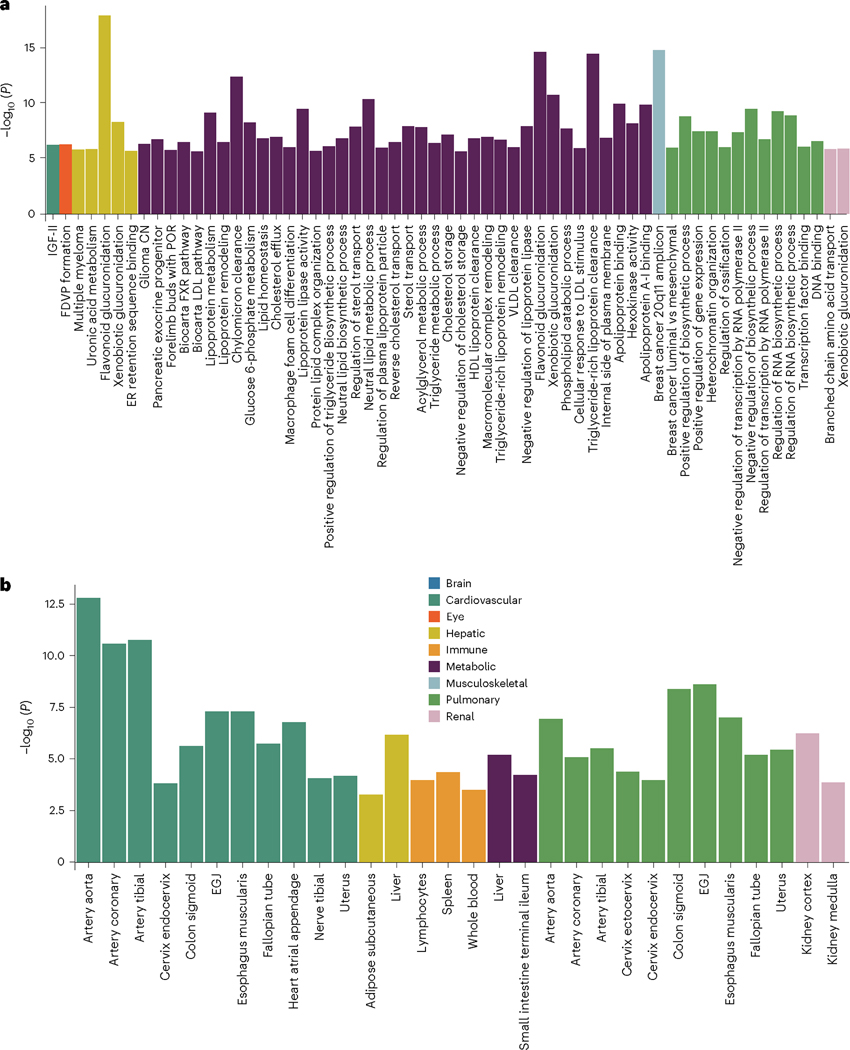
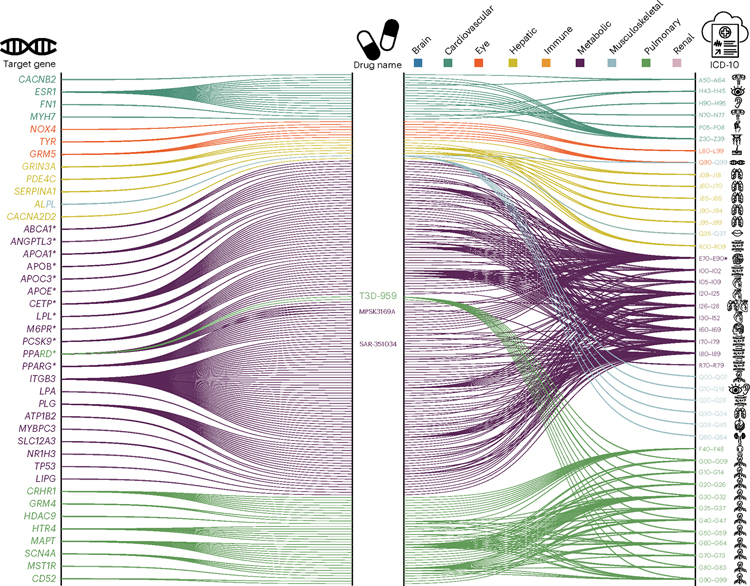
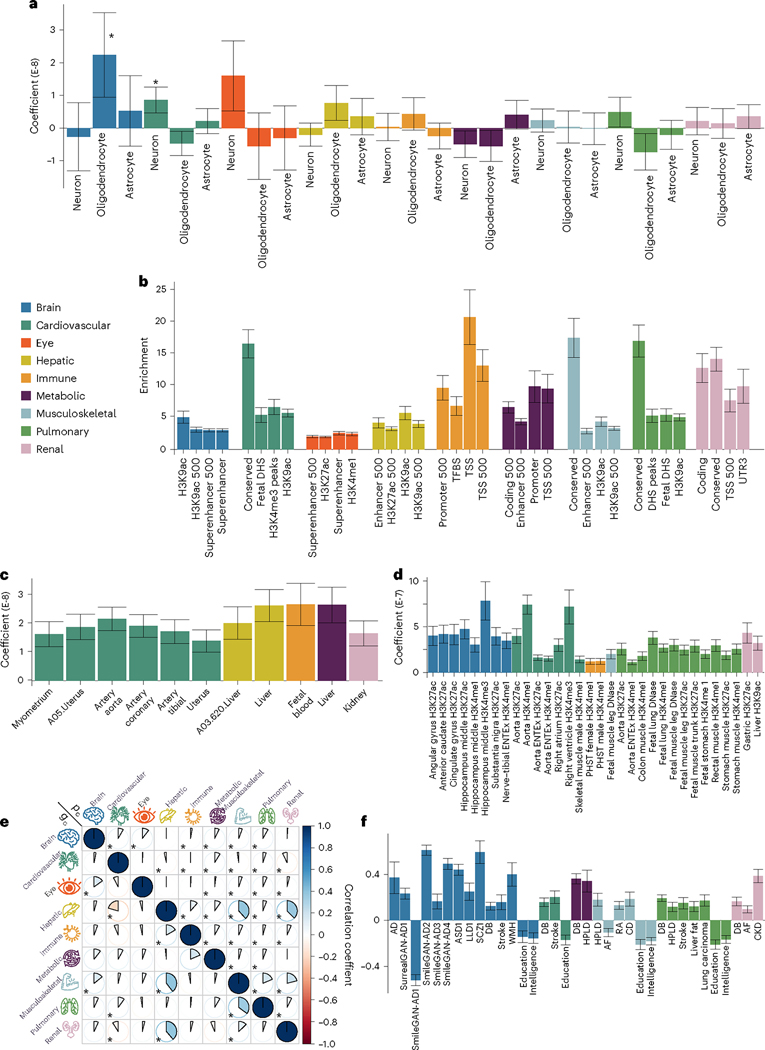
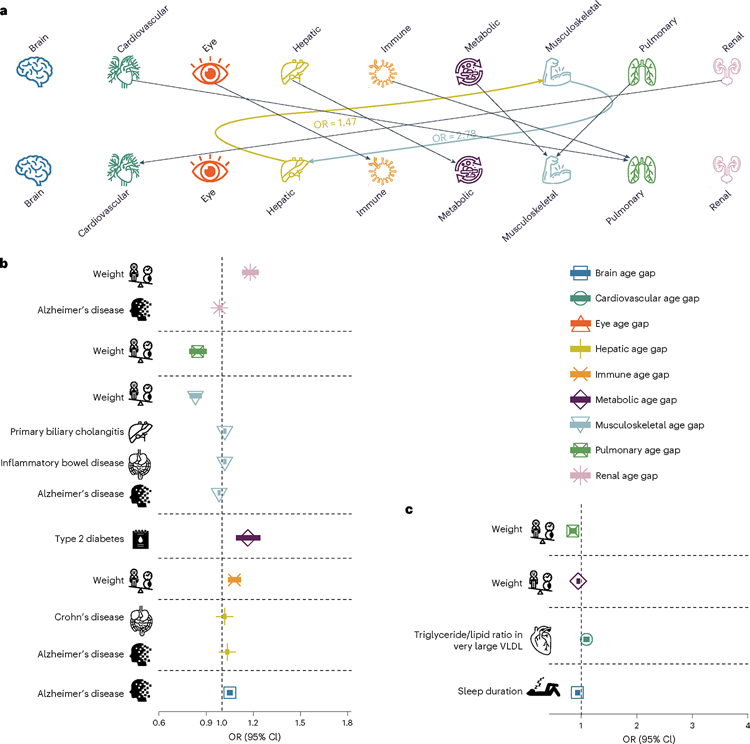
Investigating the genetic underpinnings of human aging is essential for unraveling the etiology of and developing actionable therapies for chronic diseases. Here, we characterize the genetic architecture of the biological age gap (BAG; the difference between machine learning-predicted age and chronological age) across nine human organ systems in 377,028 participants of European ancestry from the UK Biobank. The BAGs were computed using cross-validated support vector machines, incorporating imaging, physical traits and physiological measures. We identify 393 genomic loci-BAG pairs (P < 5 × 10-8) linked to the brain, eye, cardiovascular, hepatic, immune, metabolic, musculoskeletal, pulmonary and renal systems. Genetic variants associated with the nine BAGs are predominantly specific to the respective organ system (organ specificity) while exerting pleiotropic links with other organ systems (interorgan cross-talk). We find that genetic correlation between the nine BAGs mirrors their phenotypic correlation. Further, a multiorgan causal network established from two-sample Mendelian randomization and latent causal variance models revealed potential causality between chronic diseases (for example, Alzheimer's disease and diabetes), modifiable lifestyle factors (for example, sleep duration and body weight) and multiple BAGs. Our results illustrate the potential for improving human organ health via a multiorgan network, including lifestyle interventions and drug repurposing strategies.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Update of
-
The Genetic Architecture of Biological Age in Nine Human Organ Systems.medRxiv [Preprint]. 2024 Jun 11:2023.06.08.23291168. doi: 10.1101/2023.06.08.23291168. medRxiv. 2024. Update in: Nat Aging. 2024 Sep;4(9):1290-1307. doi: 10.1038/s43587-024-00662-8. PMID: 37398441 Free PMC article. Updated. Preprint.
References
-
- Hodson R Precision medicine. Nature 537, S49 (2016). - PubMed
-
- Wen J et al. Genetic, clinical underpinnings of brain change along two neuroanatomical dimensions of clinically-defined Alzheimer’s disease. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/2022.09.16.508329v3 (2024). - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
