

Assessing the impact of transcriptomics data analysis pipelines on downstream functional enrichment results
- PMID: 38943333
- PMCID: PMC11317128
- DOI: 10.1093/nar/gkae552
Assessing the impact of transcriptomics data analysis pipelines on downstream functional enrichment results
Abstract
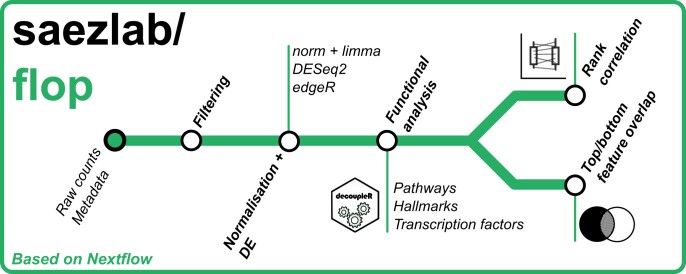
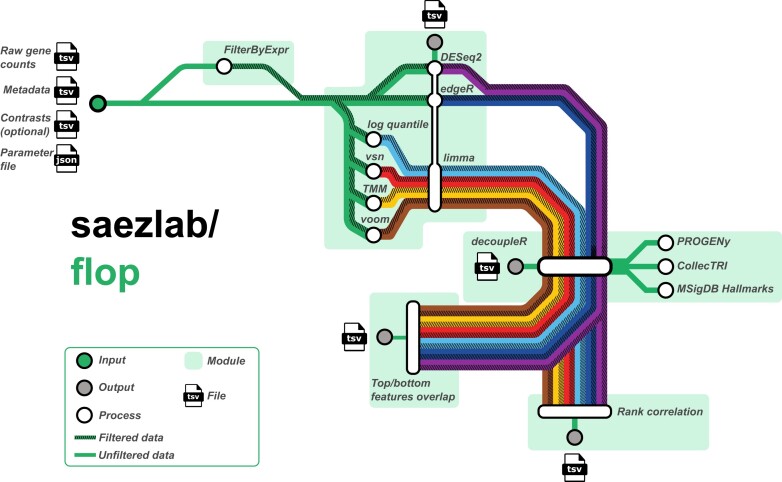
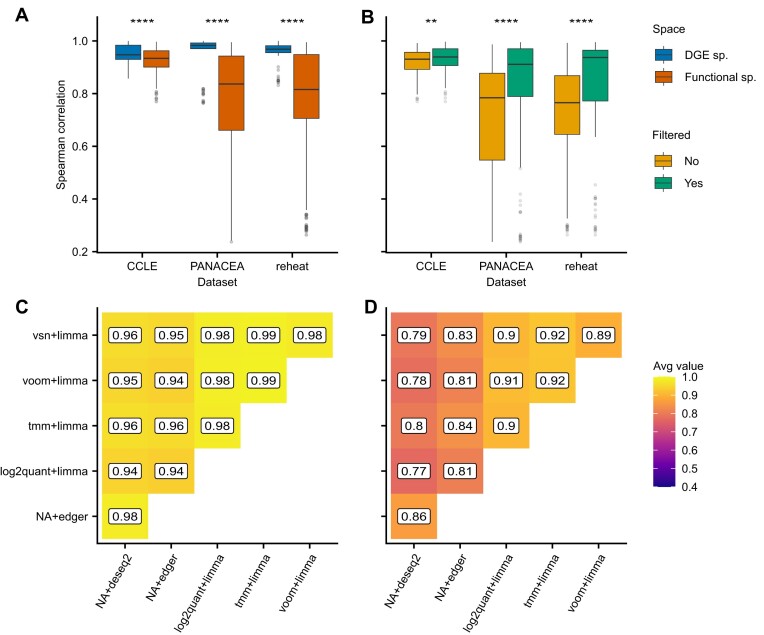
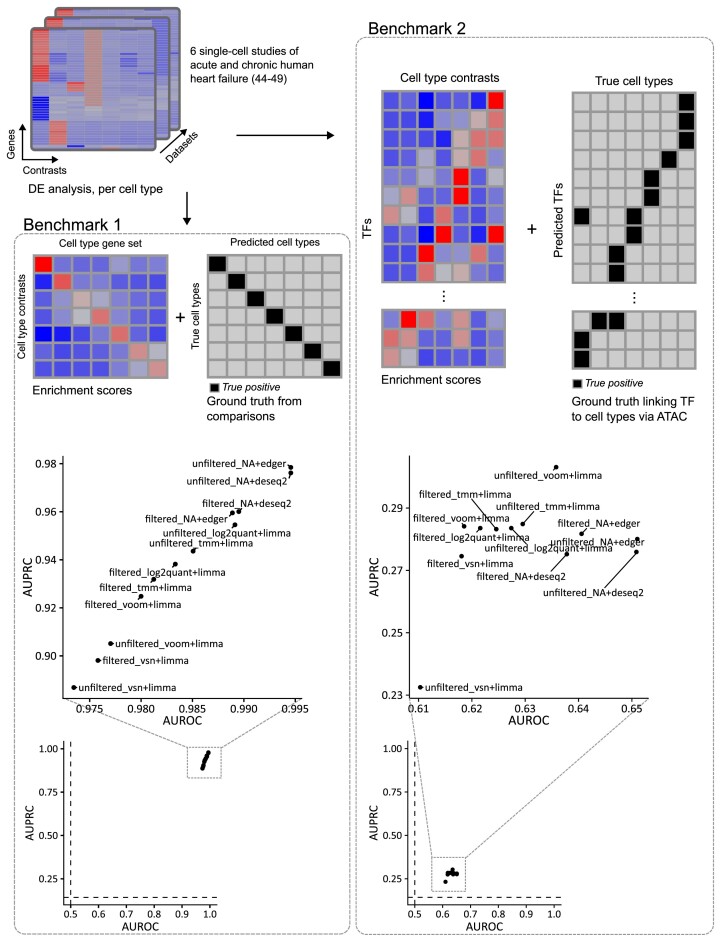
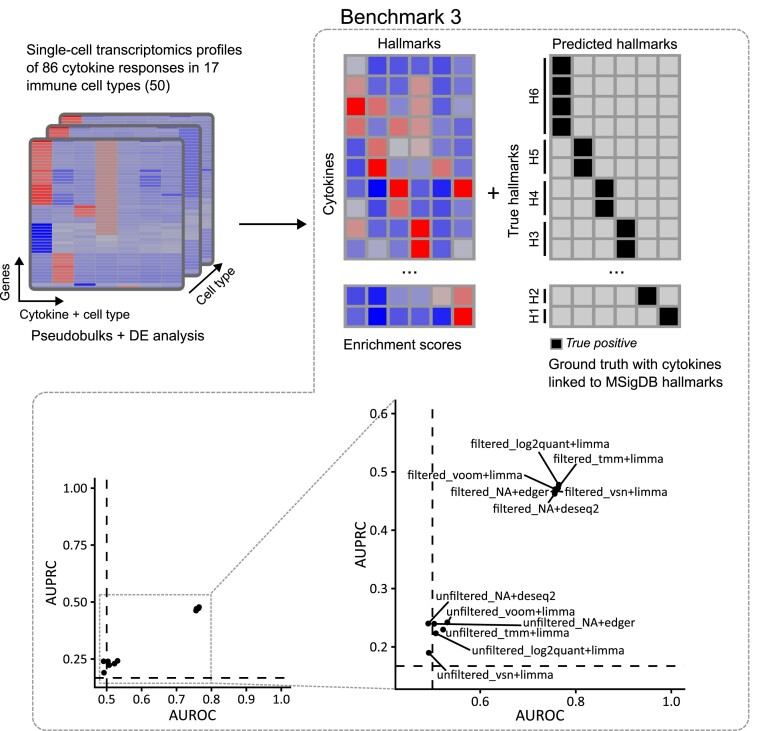
Transcriptomics is widely used to assess the state of biological systems. There are many tools for the different steps, such as normalization, differential expression, and enrichment. While numerous studies have examined the impact of method choices on differential expression results, little attention has been paid to their effects on further downstream functional analysis, which typically provides the basis for interpretation and follow-up experiments. To address this, we introduce FLOP, a comprehensive nextflow-based workflow combining methods to perform end-to-end analyses of transcriptomics data. We illustrate FLOP on datasets ranging from end-stage heart failure patients to cancer cell lines. We discovered effects not noticeable at the gene-level, and observed that not filtering the data had the highest impact on the correlation between pipelines in the gene set space. Moreover, we performed three benchmarks to evaluate the 12 pipelines included in FLOP, and confirmed that filtering is essential in scenarios of expected moderate-to-low biological signal. Overall, our results underscore the impact of carefully evaluating the consequences of the choice of preprocessing methods on downstream enrichment analyses. We envision FLOP as a valuable tool to measure the robustness of functional analyses, ultimately leading to more reliable and conclusive biological findings.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
