

Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank
- PMID: 38944039
- PMCID: PMC11293579
- DOI: 10.1016/j.xgen.2024.100602
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank
Abstract
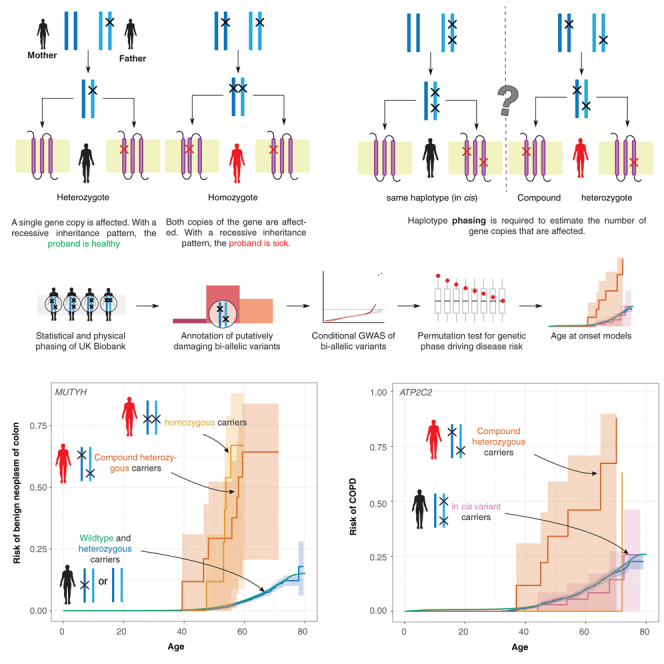
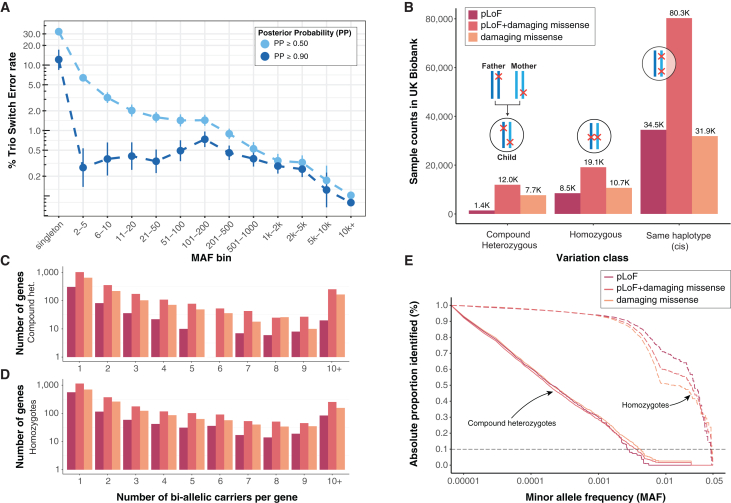
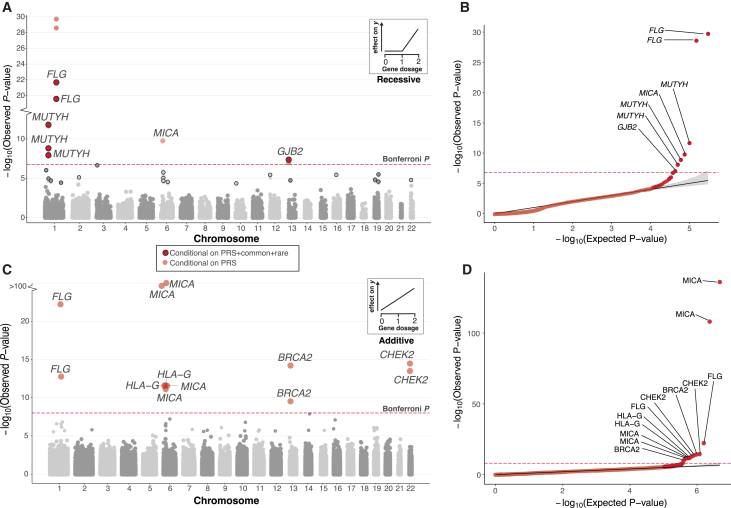
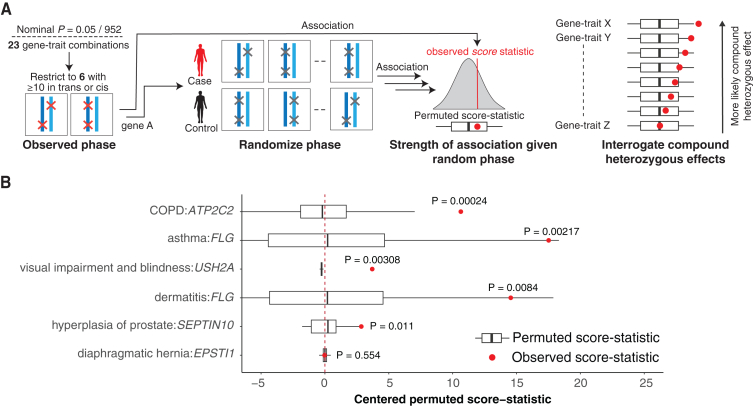
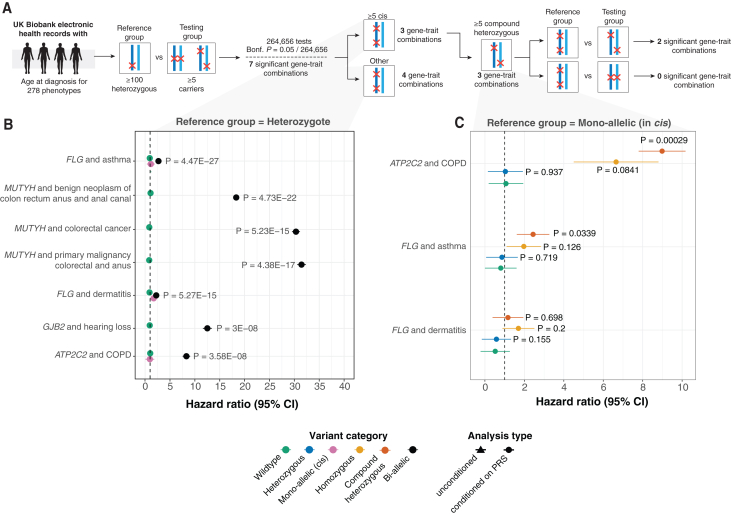
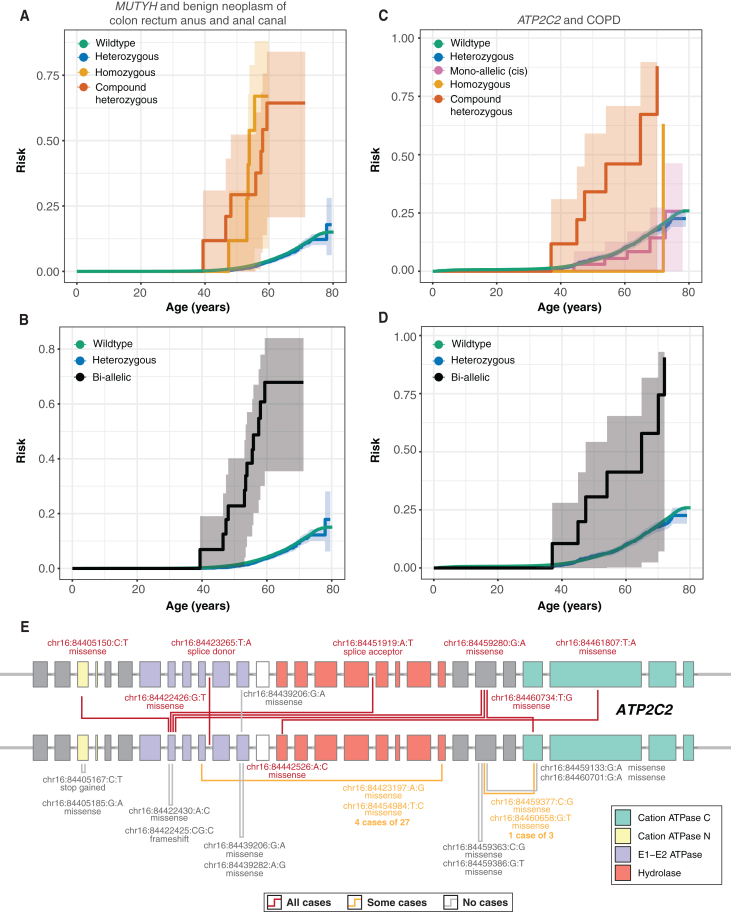
The phenotypic impact of compound heterozygous (CH) variation has not been investigated at the population scale. We phased rare variants (MAF ∼0.001%) in the UK Biobank (UKBB) exome-sequencing data to characterize recessive effects in 175,587 individuals across 311 common diseases. A total of 6.5% of individuals carry putatively damaging CH variants, 90% of which are only identifiable upon phasing rare variants (MAF < 0.38%). We identify six recessive gene-trait associations (p < 1.68 × 10-7) after accounting for relatedness, polygenicity, nearby common variants, and rare variant burden. Of these, just one is discovered when considering homozygosity alone. Using longitudinal health records, we additionally identify and replicate a novel association between bi-allelic variation in ATP2C2 and an earlier age at onset of chronic obstructive pulmonary disease (COPD) (p < 3.58 × 10-8). Genetic phase contributes to disease risk for gene-trait pairs: ATP2C2-COPD (p = 0.000238), FLG-asthma (p = 0.00205), and USH2A-visual impairment (p = 0.0084). We demonstrate the power of phasing large-scale genetic cohorts to discover phenome-wide consequences of compound heterozygosity.
Keywords: bi-allelic; compound heterozygosity; longtudinal; phasing; population genetics; recessive.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.M.N. is a member of the scientific advisory board at Deep Genomics and Neumora.
Figures
Update of
-
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank.medRxiv [Preprint]. 2023 Jul 3:2023.06.29.23291992. doi: 10.1101/2023.06.29.23291992. medRxiv. 2023. Update in: Cell Genom. 2024 Jul 10;4(7):100602. doi: 10.1016/j.xgen.2024.100602. PMID: 37461573 Free PMC article. Updated. Preprint.
Similar articles
-
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank.medRxiv [Preprint]. 2023 Jul 3:2023.06.29.23291992. doi: 10.1101/2023.06.29.23291992. medRxiv. 2023. Update in: Cell Genom. 2024 Jul 10;4(7):100602. doi: 10.1016/j.xgen.2024.100602. PMID: 37461573 Free PMC article. Updated. Preprint.
-
Analysis of Rare Coding Variants in 470,000 UK Biobank Participants Reveals Genetic Associations With Childhood Asthma Predisposition.Int J Immunogenet. 2025 Jun;52(3):155-161. doi: 10.1111/iji.12714. Epub 2025 May 8. Int J Immunogenet. 2025. PMID: 40342259
-
Investigation of Recessive Effects of Coding Variants on Common Clinical Phenotypes in Exome-Sequenced UK Biobank Participants.Hum Hered. 2024;89(1):1-7. doi: 10.1159/000537771. Epub 2024 Feb 10. Hum Hered. 2024. PMID: 38342085
-
Exome sequencing and analysis of 454,787 UK Biobank participants.Nature. 2021 Nov;599(7886):628-634. doi: 10.1038/s41586-021-04103-z. Epub 2021 Oct 18. Nature. 2021. PMID: 34662886 Free PMC article.
-
Rare variant contribution to human disease in 281,104 UK Biobank exomes.Nature. 2021 Sep;597(7877):527-532. doi: 10.1038/s41586-021-03855-y. Epub 2021 Aug 10. Nature. 2021. PMID: 34375979 Free PMC article.
Cited by
-
Saturation mapping of MUTYH variant effects using DNA repair reporters.bioRxiv [Preprint]. 2025 Mar 6:2025.03.01.640912. doi: 10.1101/2025.03.01.640912. bioRxiv. 2025. Update in: Am J Hum Genet. 2025 Sep 4;112(9):2010-2026. doi: 10.1016/j.ajhg.2025.07.005. PMID: 40093110 Free PMC article. Updated. Preprint.
-
Inferring compound heterozygosity from large-scale exome sequencing data.Nat Genet. 2024 Jan;56(1):152-161. doi: 10.1038/s41588-023-01608-3. Epub 2023 Dec 6. Nat Genet. 2024. PMID: 38057443 Free PMC article.
-
Inferring compound heterozygosity from large-scale exome sequencing data.bioRxiv [Preprint]. 2023 Aug 21:2023.03.19.533370. doi: 10.1101/2023.03.19.533370. bioRxiv. 2023. Update in: Nat Genet. 2024 Jan;56(1):152-161. doi: 10.1038/s41588-023-01608-3. PMID: 36993580 Free PMC article. Updated. Preprint.
-
Genome-wide association testing beyond SNPs.Nat Rev Genet. 2025 Mar;26(3):156-170. doi: 10.1038/s41576-024-00778-y. Epub 2024 Oct 7. Nat Rev Genet. 2025. PMID: 39375560 Free PMC article. Review.
References
-
- Nelson M.R., Tipney H., Painter J.L., Shen J., Nicoletti P., Shen Y., Floratos A., Sham P.C., Li M.J., Wang J., et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015;47:856–860. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
